
Phase 3
 Objective
Objective
To establish the non-inferiority of oral Levonadifloxacin (Alalevonadifloxacin) with oral linezolid and the non-inferiority of IV Levonadifloxacin with IV linezolid in ABSSSI at the Test of Cure (TOC) visit.
 Methodology
Methodology
The study design and methodology used are as per the USFDA guideline.
 Study design
Study design
This was a Phase 3, multi-centre, randomized, active-comparator study in subjects with ABSSSI. The study had two subgroups for assessment of efficacy and safety (oral subgroup 1 and IV subgroup 2).
Eligibility for inclusion to the oral or IV subgroup was determined by the investigator. Within each subgroup (IV or Oral), subjects were randomized to Levonadifloxacin/Alalevonadifloxacin or linezolid using a 1:1 allocation ratio.
In each subgroup, 250 subjects (125/arm) were randomized so that the study had sufficient power to meet its objectives.
 Subjects
Subjects
Inclusion criteria: Eligibility criteria were age 18 to 65 years and a diagnosis of ABSSSI classified as cellulitis/erysipelas, wound infection or major cutaneous abscess with lesion size area of at least 75 cm2 and suspected and/or documented evidence of Gram-positive infection
Exclusion criteria: Receipt of systemic antibiotic therapy within the past 24 hours before enrolment unless one of the following was documented - subjects with evidence of clinical progression of ABSSSI while on antibacterial drug therapy after at least 48 hours, subjects who received a single dose of a short-acting antibacterial drug within 24 hours of enrolment and subjects who received an antibacterial drug for surgical prophylaxis and subsequently developed ABSSSI.
Subjects who were willing to participate in the study and fulfilled all eligibility criteria were enrolled at 32 study centers across India between August 2017 and August 2018.
Subjects received either oral therapy (subgroup 1) or IV therapy (subgroup 2) and were randomized (1:1) to Levonadifloxacin/Alalevonadifloxacin or linezolid within each subgroup with equal numbers (N=250) enrolled in the oral & IV subgroups for a total of 500 planned subjects.
Linezolid was chosen as the comparator as it is one of very few available options for the treatment of MRSA infections with both oral as well as parenteral formulations.
 Interventions
Interventions
Subjects received initial treatment with either oral or IV therapy based on clinical judgment of the investigator.
Subjects who received treatment with oral therapy were randomized to receive either oral Levonadifloxacin 1000 mg twice daily or oral linezolid 600 mg twice daily for a treatment period of 7 to 10 days.
Subjects who received initial treatment with IV therapy were randomized to receive either IV Levonadifloxacin 800 mg twice daily or IV linezolid 600 mg twice daily.
For subjects who received initial therapy by parenteral route, investigators were allowed to switch to oral therapy after at least 48 hours of IV treatment as per their discretion, provided there was a reduction in lesion size or no increase in lesion size from baseline, and no new loci of infection.
Cumulative treatment duration (IV and Oral) remained 7 to 10 days. All subjects received IV aztreonam (1 g twice daily) for Gram-negative coverage starting from the day of randomization.
If the baseline culture identified aztreonam-resistant Gram-negative strains and the subject was non-responder, IV colistin 4 million IU twice daily was initiated.
Blood samples were collected from approximately 25% of subjects in Alalevonadifloxacin/Levonadifloxacin treatment arms (oral and IV) to determine the plasma concentrations of Levonadifloxacin and its sulphate metabolite.
 Study visits
Study visits
At screening, randomization on Day 1, twice on therapy visits on Day 3 or 4 and Day 7 or 8, End of Therapy (EOT) Visit (performed within three days of the last dose of study medication), Test of Cure (TOC) Visit (conducted 7 to 14 days after administration of the last dose of study medication) and an all-cause mortality assessment (performed between Day 28 to Day 35 of the study, which could also have been performed telephonically).
 Efficacy assessments
Efficacy assessments
The primary outcome measure of the study was the overall clinical response at the TOC Visit in the modified intent-to-treat (mITT) population, separately for the Oral and IV subgroups.
The secondary clinical outcomes of the study were the clinical response at Visit 3 in the mITT population, the overall clinical response at the EOT Visit in the mITT and the clinically evaluable intent-to-treat (CE-ITT) populations, the overall clinical response at the TOC Visit in the CE-ITT population, and the microbiological response at the EOT and TOC Visits in the microbiological intent-to-treat (micro-ITT) and the microbiologically evaluable intent-to-treat (ME-ITT) populations.
Clinical Response at Day 3 was determined using the percentage reduction in lesion size (measured by the area of redness, oedema, or induration).
Subjects with a percent reduction greater than or equal to 20% compared to baseline were considered clinical responders at Day 3.
Subjects meeting any of the following criteria – a) less than 20% reduction in lesion size compared to baseline, b) increase in lesion size compared to baseline, c) receipt of rescue therapy before Visit 3 or d) not alive, were considered clinical non-responders at Day 3.
Subjects missing data necessary to determine a treatment response at Day 3 were deemed to have a clinical indeterminate outcome.
Overall Clinical Response (at EOT and also at the TOC visit) was considered Clinical Cure if there was resolution of all clinical signs and symptoms of ABSSSI infection.
Overall Clinical Response were considered Clinical Failure, if there was nonresolution of clinical signs and symptoms of infection or receipt of rescue therapy or death. Overall Clinical Response were considered Indeterminate in cases with missing data.
 Microbiological assessments
Microbiological assessments
Microbiological response at EOT and TOC for subjects in the microbiological intent-to-treat (micro-ITT) and the microbiologically evaluable intent-to-treat (ME-ITT) population was based on the collection of an adequate deep specimen obtained from the infection site for Gram staining, culture and susceptibility testing.
The sample for blood and microbiological culture was collected at the Screening or Randomization Visit (if not collected at the Screening Visit) and Subjects were evaluated for microbiological response at the EOT and TOC Visits.
Microbiological response was categorized as: Documented eradicated (baseline pathogen absent in post treatment cultures); presumed eradicated (no post treatment material available for culture with a clinical response of success); documented persisted (baseline pathogen present in post treatment cultures); or presumed persisted (no post treatment material available for culture with an investigator-assessed response of clinical failure).
 Safety and tolerability assessments
Safety and tolerability assessments
Safety evaluation was based on adverse events reported, vital signs, physical examination findings, clinical laboratory evaluation, and ECG collected during the study.
Descriptive summaries (frequencies and percentages) for specific TEAEs were presented by system organ class and preferred term according to the Medical Dictionary for Regulatory Activities (MedDRA v19.1) dictionary by treatment group and also for the pooled treatment group (IV and oral).
 Statistical analysis
Statistical analysis
The primary efficacy analysis was based on the mITT population. Secondary efficacy analyses of the clinical outcomes were based on the mITT, and CE-ITT populations.
Microbiological outcomes (subject level and per-pathogen) were conducted for the micro-ITT and ME-ITT populations.
All efficacy analyses were conducted according to the randomized treatment assignment (ITT population were all subjects who were randomized, mITT population were all subjects who were randomized, received at least one dose of study drug and had at least one post-baseline efficacy measurement, Micro-ITT population were all subjects randomized to treatment who have a baseline Gram-positive bacterial pathogen (including MRSA).
Clinically evaluable (CE-ITT) population were ITT subjects who followed important components of the trial and microbiologically evaluable intent-to-treat (ME-ITT) populations were micro-ITT subjects who followed important components of the trial).
There were two co-primary comparisons to evaluate the non-inferiority of Levonadifloxacin versus linezolid:
1. Oral Levonadifloxacin (T1) versus oral linezolid (C1)
2. IV Levonadifloxacin (T2) versus IV linezolid (C2)
Non-inferiority was assessed by comparing the lower limit of the 95% confidence interval for the difference in overall clinical cure rates at TOC (T1-C1 and T2-C2) in the mITT population to the non-inferiority margin of 15%. The 15% margin was chosen based on historical studies of the active controls with placebo.
If non-inferiority was demonstrated for both the comparisons (1) and (2), then the primary objective of the study was considered to be met.
All PK and safety endpoints were assessed using appropriate descriptive statistical summaries.
 Results
Results
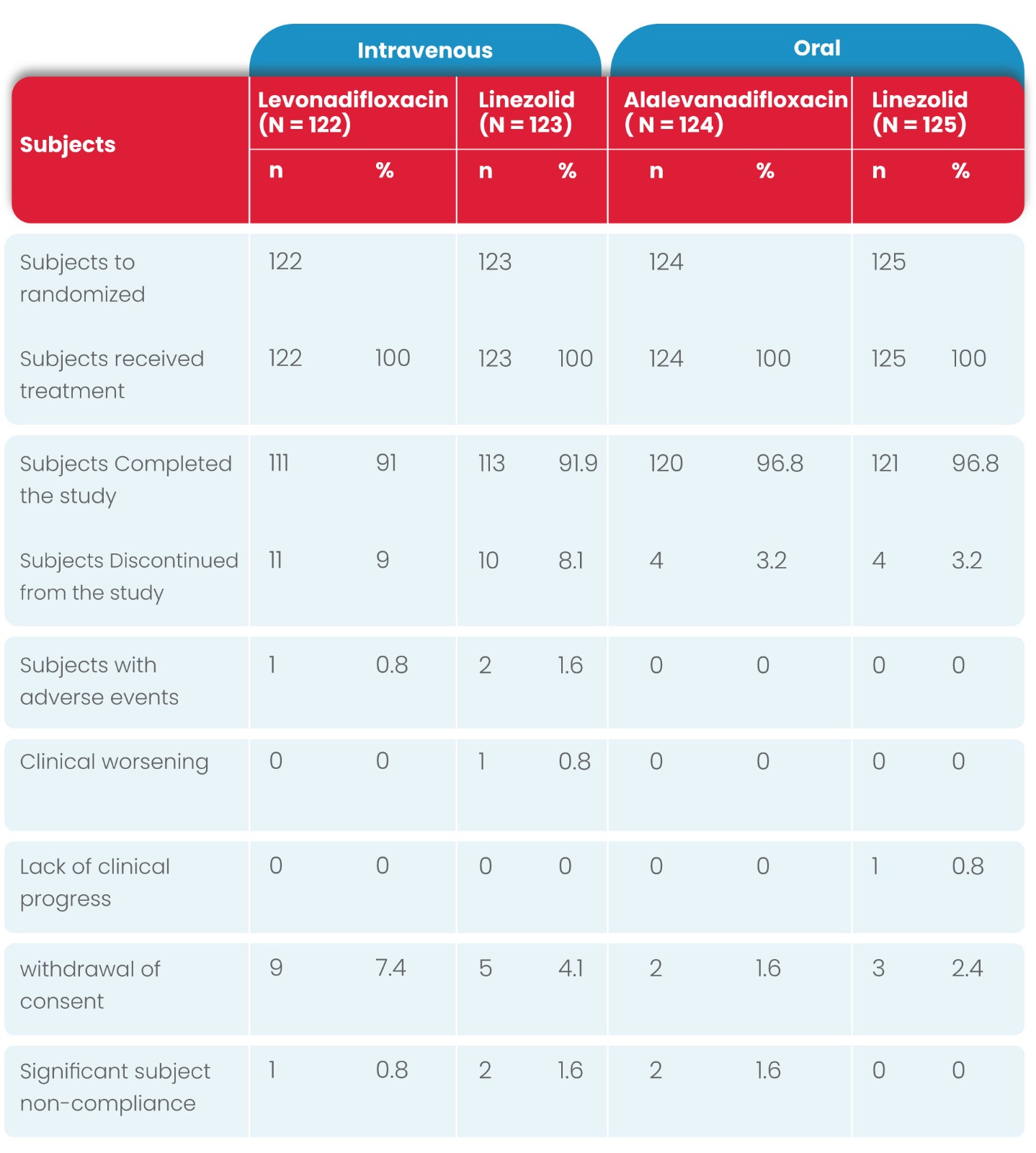
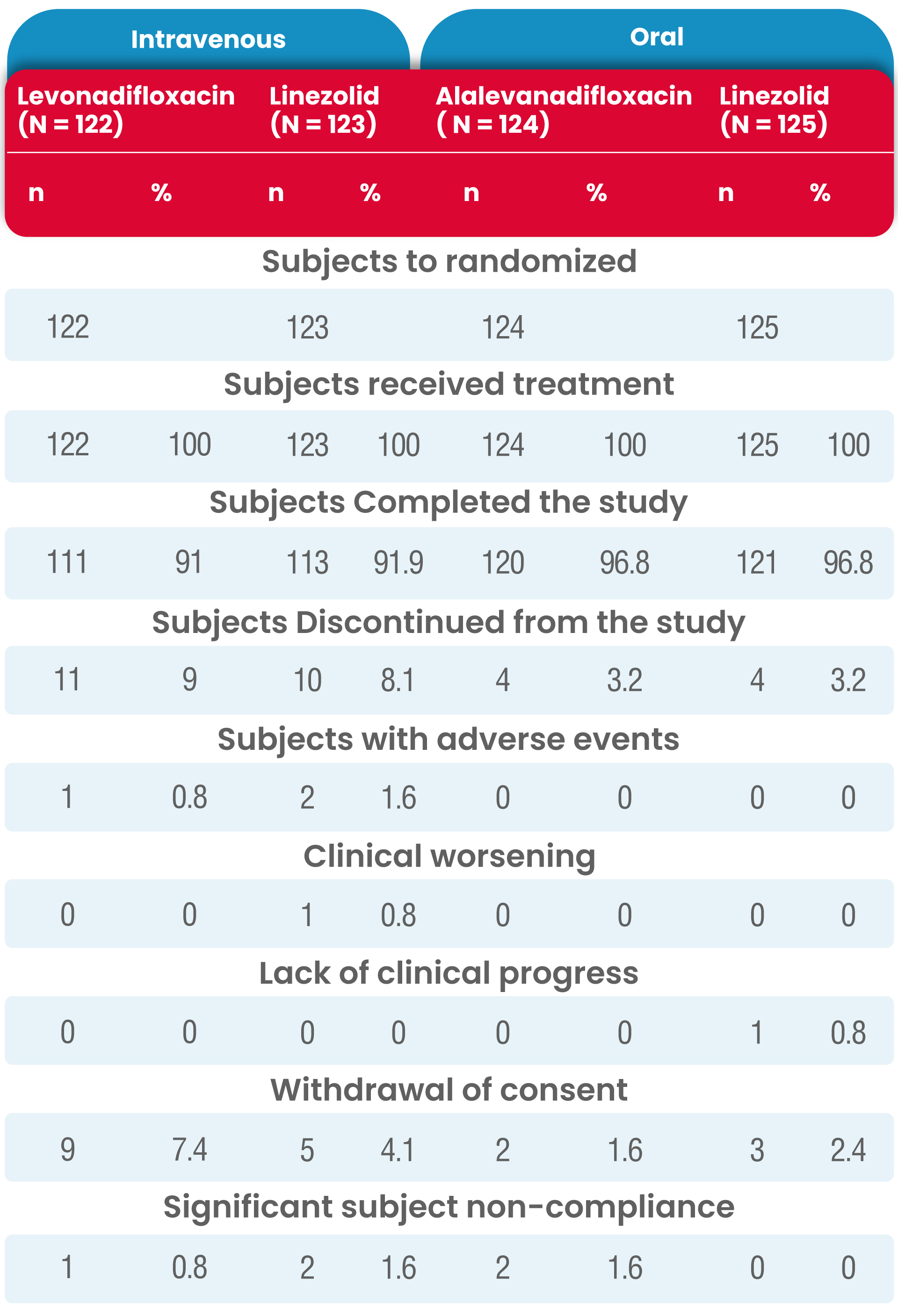
Subject disposition and analysis sets
A total of 501 subjects (IV subgroup: 250; oral subgroup: 251) were randomized, 465 subjects completed the study while 36 subjects discontinued early from the study.
Similar disposition was reported in the Mitt population for both subgroups as summarized in the following table. The number of subjects in each population (Mitt, micro-ITT, CE-ITT, and ME-ITT) were similar between the two treatment groups.
Subject demographic and clinical characteristics
All subjects were of the Indian race. Overall the majority of the subjects were male, with a mean age of approximately 45 years and average BMI range of 18.5 to <25 kg/m2.
In the majority of subjects, the pain and tenderness at the wound site was considered moderate to severe grade at baseline.
The site of infection was the leg in approximately half of the subjects followed by approximately 20% with a foot infection.
The majority of the subjects had a diagnosis of cellulitis/erysipelas or wound infection. The incidence of cellulitis/erysipelas was higher in the oral subgroup compared to the IV subgroup (45% vs. 37.1%, respectively); whereas the incidence of wound infections was higher in the IV subgroup (39.6% vs. 32.1%).
The mean lesion size (area) was higher in the subjects receiving IV therapy compared to the subjects receiving oral therapy (174.27 vs.130.38 cm2, respectively).
However, the mean lesion size was similar within each of the two treatment groups (IV and oral).
The baseline characteristics were comparable between the treatment groups and between the IV and oral subgroups.
Diabetic foot ulcer was reported in approximately 10% of subjects in the IV subgroup and 5% of subjects in the oral subgroup at baseline.
Bacteraemia was reported in eight subjects (four in Levonadifloxacin; four in linezolid) in the IV subgroup, compared to four subjects (one in Levonadifloxacin; three in linezolid) in the oral subgroup. .
The majority of subjects had a baseline infection caused by S. aureus with approximately 30% of subjects having MRSA.
Clinical outcomes
In the mITT population, the clinical cure rates of IV and oral therapy for Levonadifloxacin were numerically higher compared to IV & oral linezolid therapy at TOC visits, respectively
In the IV and oral subgroup, 91.0% vs 87.8% and 95.2% vs 93.6% of subjects exhibited clinical cure at TOC, with a treatment difference of 3.2% and 1.6%, respectively.
As the lower limit of the 95% CI around the treatment difference was greater than -15%, non-inferiority of IV and oral Levonadifloxacin compared with IV and oral linezolid was established.
Since non-inferiority was established for each mode of administration subgroup, the primary study objective was met.
Details of the proportion of subjects whose overall clinical response outcome was a cure at TOC Visit in the mITT population is presented in the following table.
Subject disposition (Modified ITT population)
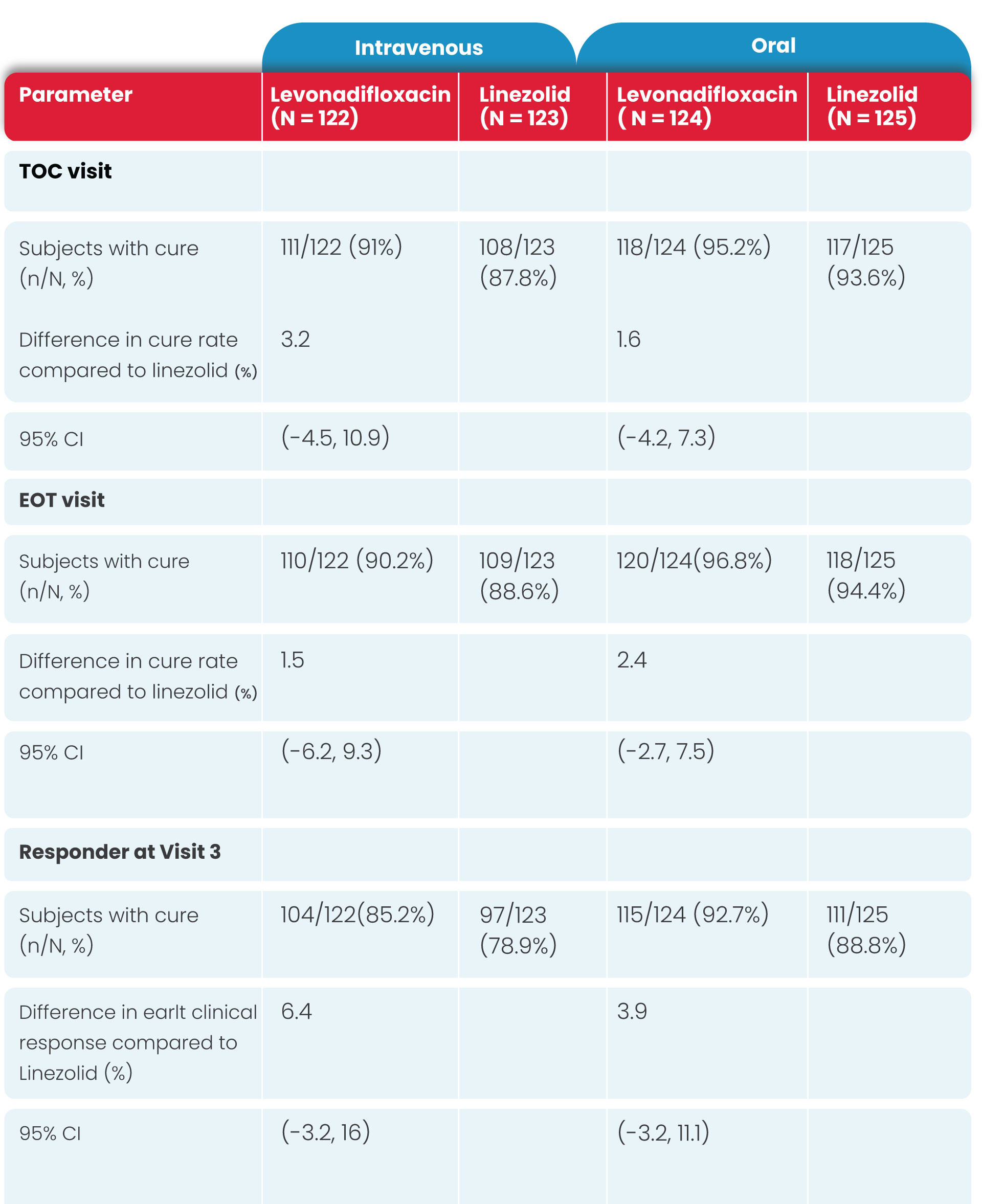
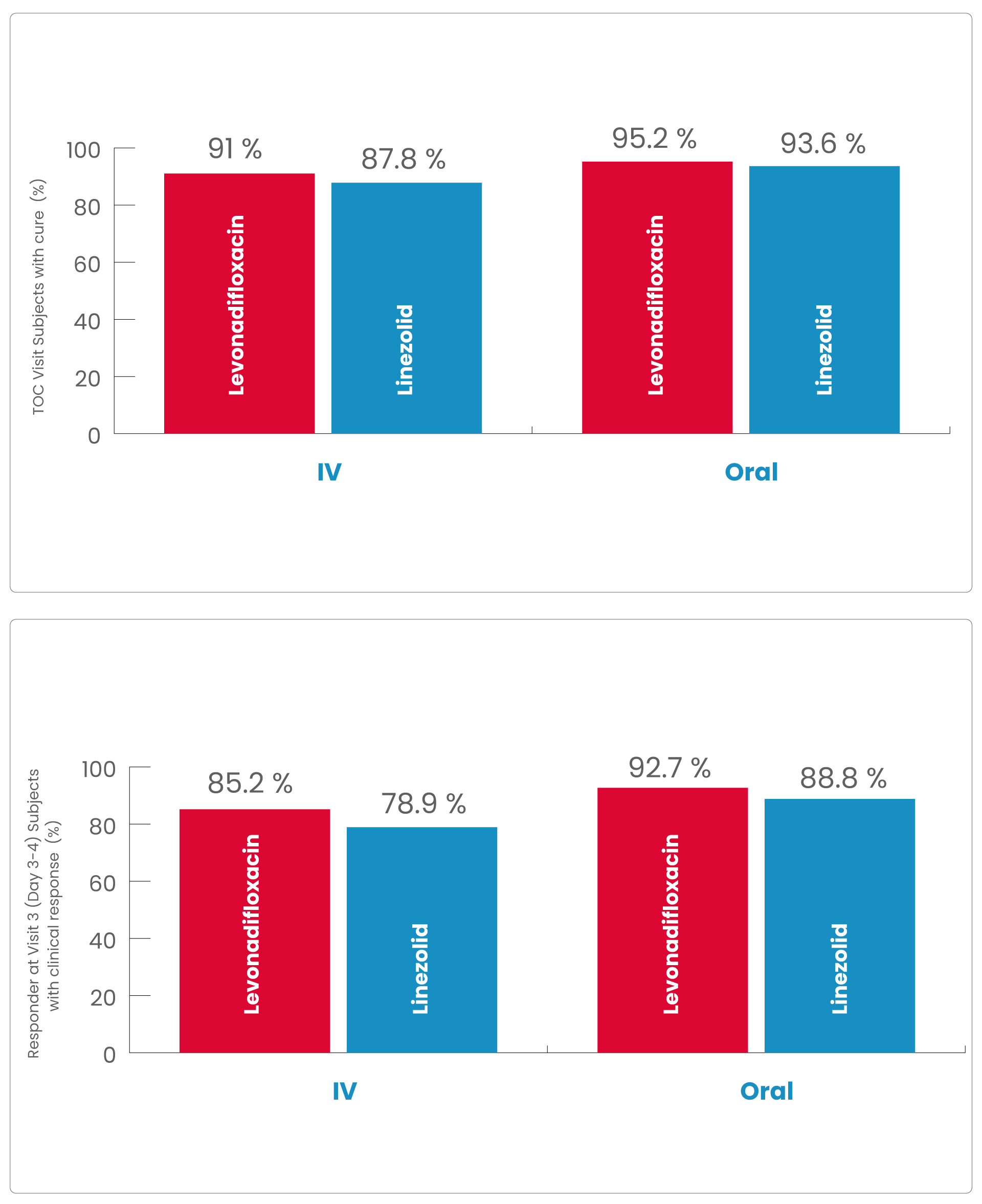
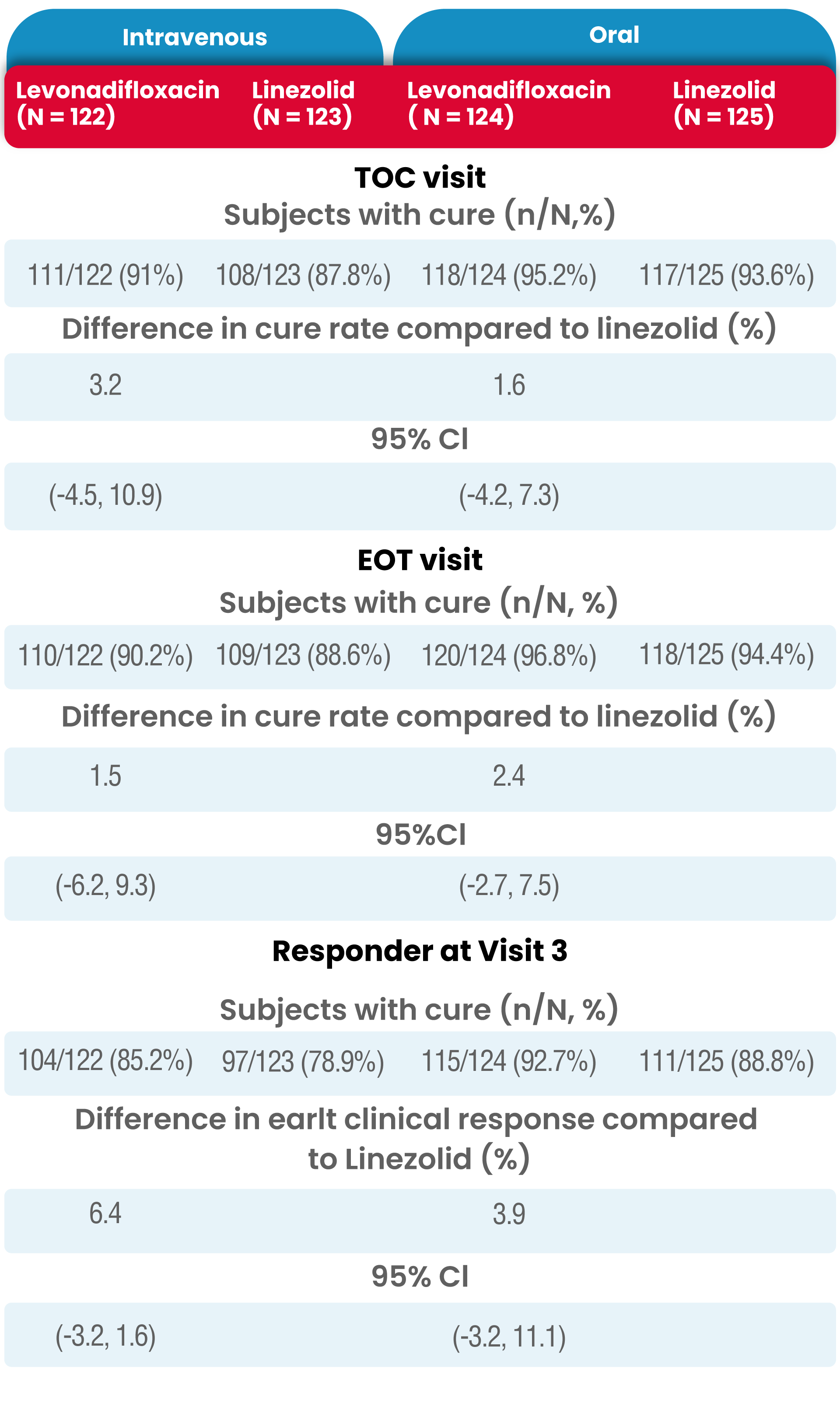
In the Levonadifloxacin treatment group, the clinical responder rate was numerically higher than in the linezolid treatment group (85.2% vs. 78.9% in IV group and 92.7% vs. 88.8% in oral group) at Visit 3 (Day 3-4) with ≥20% reduction in lesion size compared to baseline measurement, with a treatment difference of 6.4% and 3.9% in IV and oral group, respectively.
The most common reason for subjects being classified as a non-responder was <20% reduction in lesion size.
In both analysis populations (mITT and CE-ITT), the clinical cure rates at the EOT Visit was high and comparable between the two treatment (Levonadifloxacin and linezolid) groups as well as between the IV and oral subgroups.
The results of clinical response at TOC Visit in the CE-ITT population were similar to the mITT population.
Favourable clinical outcome was demonstrated in five patients having baseline Gram negative pathogens susceptible to Levonadifloxacin, but resistant to aztreonam.
There were very few clinical failures across both IV and oral treatment groups and at each visit.
The reasons for clinical failure were similar between the two treatment groups.
The most common reasons for clinical failure at the EOT visit were non-resolution of clinical signs and symptoms of infection, and receipt of rescue therapy.
Microbiological efficacy
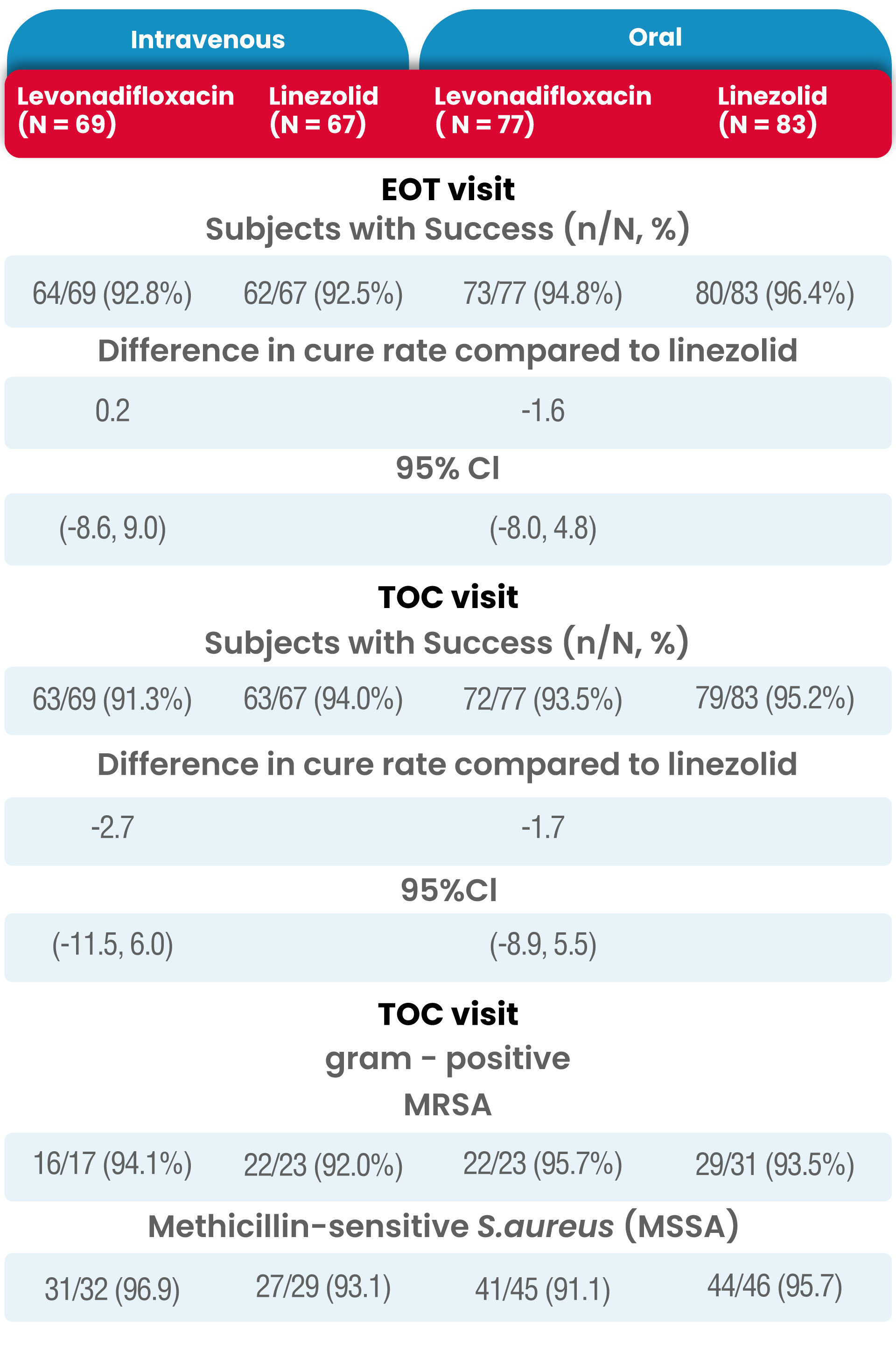
The proportion of subjects with a successful microbiological response at the EOT and TOC Visits in the micro-ITT populations is presented in Table 3.
Similar to the overall clinical response results, microbiological success (i.e., microbiological eradication and/or presumed eradication) at the EOT and TOC Visit was high and comparable between the two treatment groups.
The overall results of microbiological response rates at both the EOT and TOC Visits in the ME-ITT population (IV Levonadifloxacin: 98.2% and 94.5%, IV linezolid: 97.9% and 100%; oral Levonadifloxacin: 97.0% and 95.5%, oral linezolid: 100% and 98.6%, respectively) was similar to the high response rates observed in the micro-ITT population (IV Levonadifloxacin: 92.5% and 91.3%, IV linezolid: 92.5% and 94.0%; oral Levonadifloxacin: 94.8% and 93.5%, oral linezolid: 96.4% and 95.2%, respectively).
Levonadifloxacin (IV and oral) had a higher clinical cure rate at TOC for MRSA patients compared with linezolid (IV and oral), (95.0% vs. 89.3% respectively).
Clinical response at the visit 3, end of therapy (EOT), and Test of Cure (TOC) visit in modified ITT population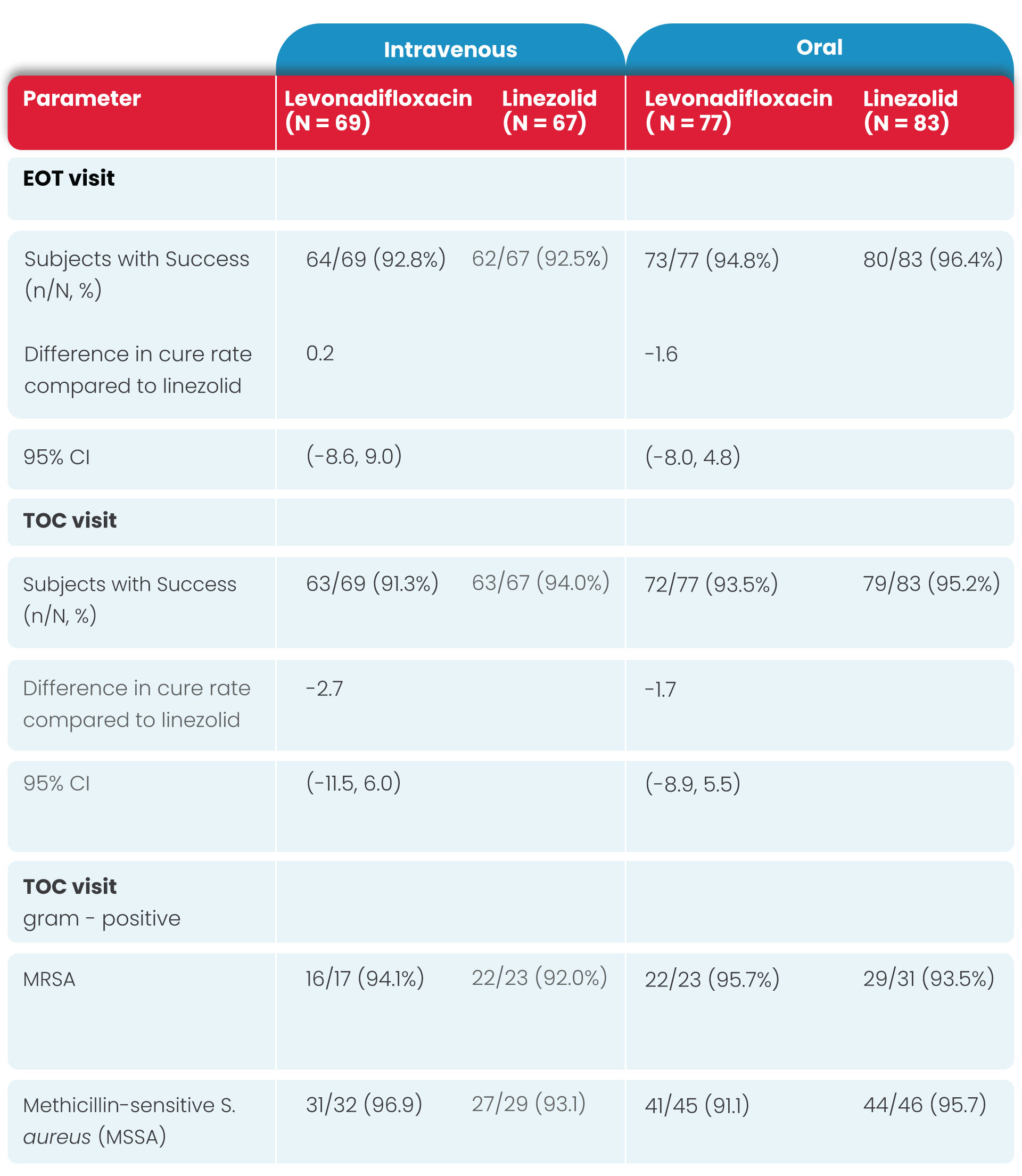
Among micro-ITT subjects, four subjects in the Levonadifloxacin IV arm (5.8%) and one each in the linezolid IV (1.5%) and oral arm (1.2%) had positive baseline blood culture, indicative of concomitant bacteraemia.
All the bacteraemia subjects in Levonadifloxacin arm, exhibited favourable clinical response as well as microbiological success where as one subject in the linezolid arm was a clinical failure despite having microbiological success
In the Diabetic Foot Ulcer sub-group, clinical cure at TOC for IV Levonadifloxacin was higher than linezolid (91.7% vs 76.9%).
Pharmacokinetic analysis
The results of the pharmacokinetic analysis showed that:
1. Levonadifloxacin, following oral and intravenous administration,presented with similar pharmacokinetic profiles (Cmax and AUC) at normalized doses
2. The bioavailability of oral Levonadifloxacin was 90%.
3. The similar pharmacokinetic profiles of Levonadifloxacin by both the IV and oral routes provide an option for IV to oral switch for the treatment of subjects.
Safety
Overall, the administration of both IV and oral Levonadifloxacin was well-tolerated in subjects with ABSSSI.
The mean treatment duration of IV (8.7 days for both Levonadifloxacin and linezolid arms) and oral therapy (8.3 days for Levonadifloxacin arm and 8.5 days for linezolid arm) were similar between the test and control drug.
Incidences of treatment-emergent adverse events (TEAEs) were similar between treatment groups and between IV (20.8% vs. 22.4%, for Levonadifloxacin and linezolid, respectively) and oral therapy (16.0% vs. 13.5% for Levonadifloxacin and linezolid, respectively).
The System Organ Class (SOC) with the highest incidence of TEAE was Gastrointestinal Disorders (5.2% vs. 6.0%, for pooled Levonadifloxacin and linezolid, respectively).
The most common AEs in Levonadifloxacin-treated subjects reported were constipation (3.6%) (with mild severity), increased blood glucose or hyperglycaemia (1.6%) (with mild to moderate severity and not related to Levonadifloxacin; the majority of these patients had high blood glucose at screening), and cough (1.2%) (with mild severity and not related to Levonadifloxacin).
The most common AEs reported in linezolid-treated subjects were vomiting (2.8%), pyrexia (2.0%), constipation (1.6%) and nausea (1.6%) as summarized in the following table.
Four subjects discontinued from the study due to TEAEs (two in the IV Levonadifloxacin group and two in the IV linezolid group).
Five serious TEAEs were reported in five subjects, all of which were in the IV treatment group.
Among them, three subjects died during the study period (one in IV Levonadifloxacin and two in IV linezolid group). All the serious TEAEs were considered not-related to the study drug(s).
Summary of treatment-emergent adverse events (safety population)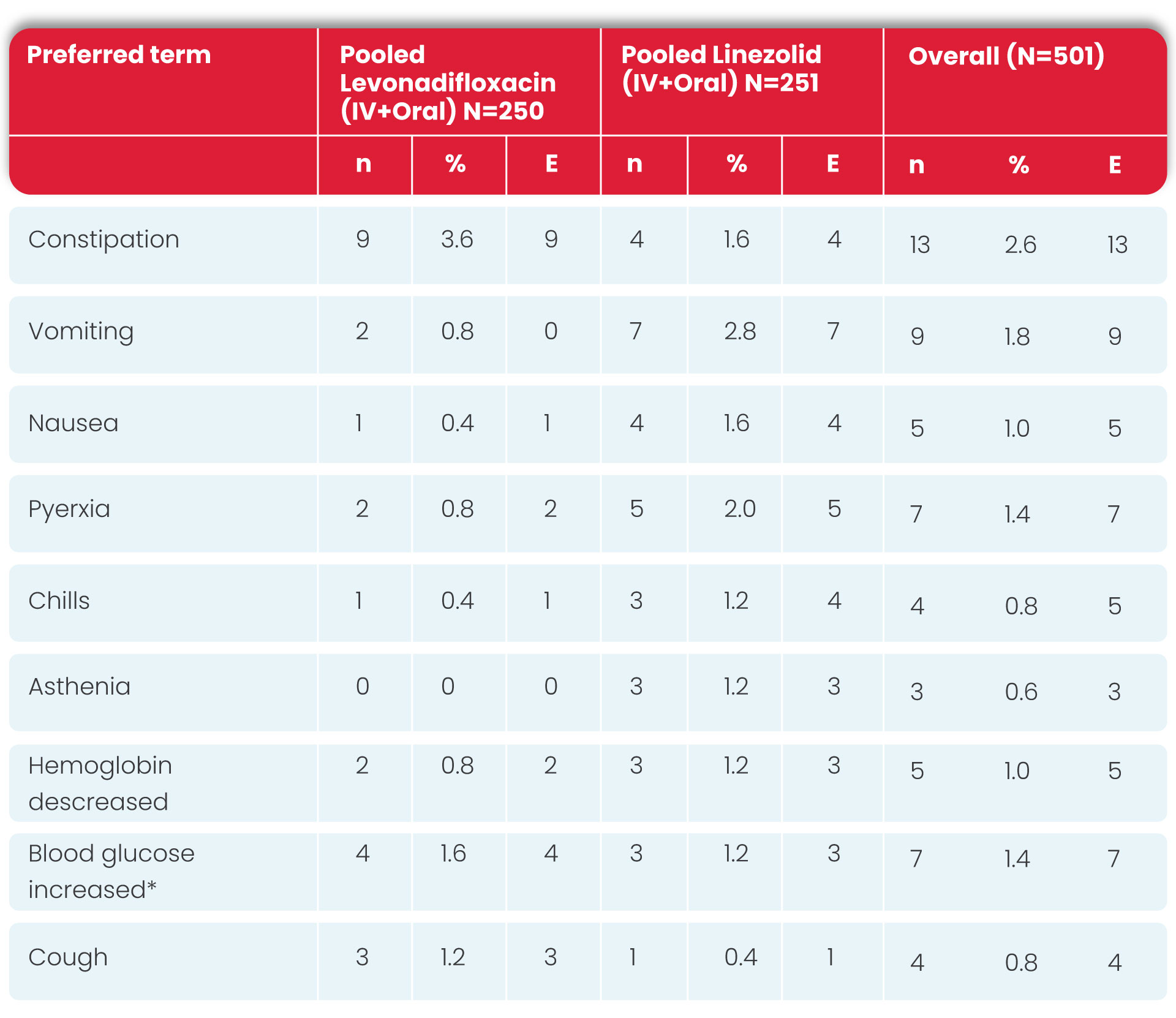
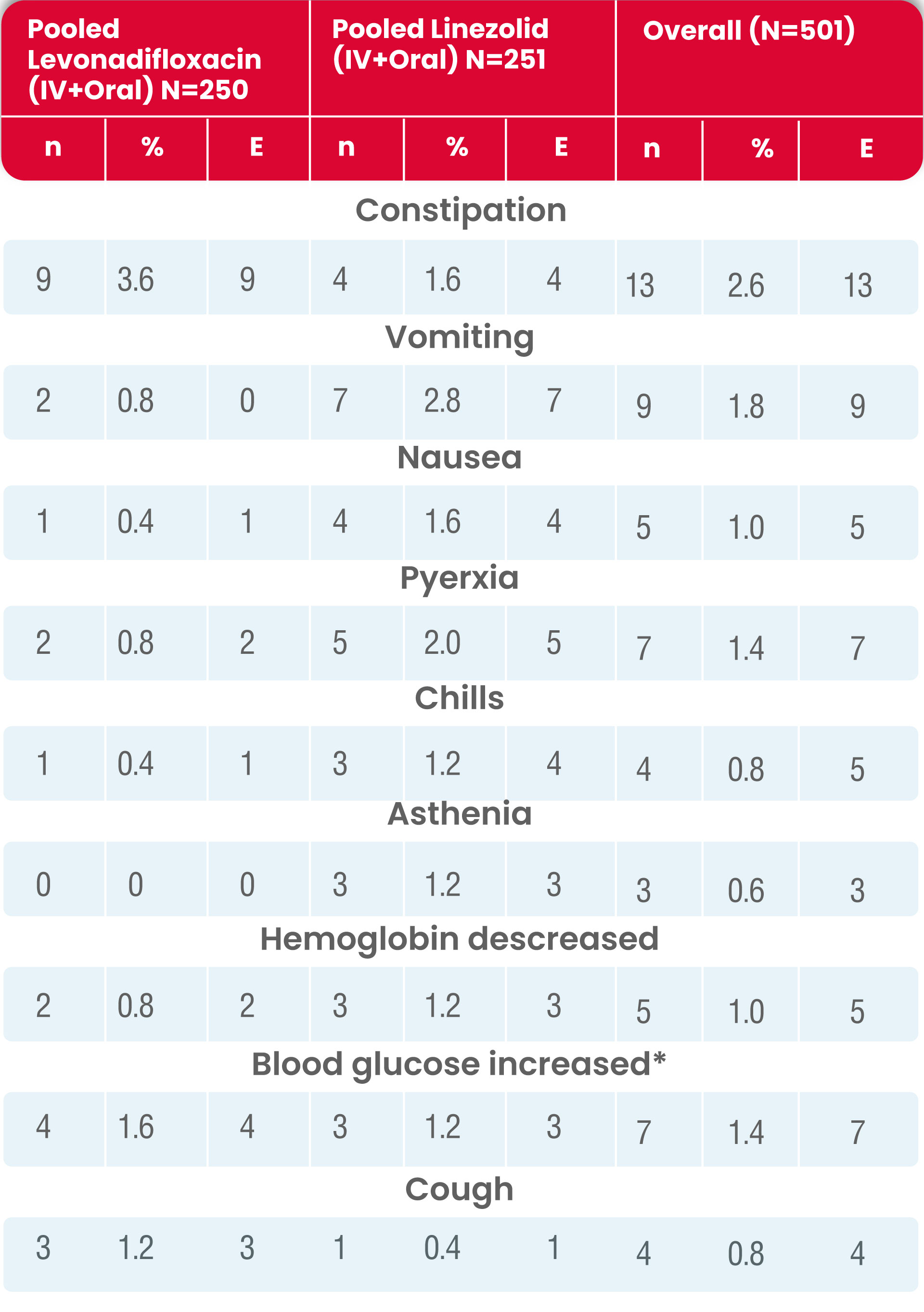
n = number of subjects with TEAE; E = number of TEAEs
* Blood glucose increase includes the cases of hyperglycemia and subjects are counted uniquely.
Major findings/outcomes of the Phase 3 trial
• Similar to previously published ABSSSI studies, the majority of the subjects enrolled in this study had an infection involving S. aureus with approximately 30% of subjects having MRSA. Cellulitis/ erysipelas and wound infections were the most common diagnosis at baseline.
• Oral and IV Levonadifloxacin (85.2% and 92.7 respectively) demonstrated high early clinical response at Visit 3 (Day 3-4).
• Clinical responses at early time points appear to be an early indicator of response and a valid approach to evaluate the effectiveness of antibacterial drugs for ABSSSIs.
• The present study also indicated that clinical response at Visit 3 is sustained and consistent at later timepoints of EOT Visit and TOC Visits.
• The microbiological success rate was high (>90%) and similar between Levonadifloxacin and linezolid at the EOT and TOC Visits in the present study.
• In micro-ITT population, the clinical cure rates of subject’s infections due to MRSA were higher for IV and oral Levonadifloxacin compared to IV and oral linezolid, respectively.
• This demonstrates that IV and oral Levonadifloxacin therapy has excellent clinical activity against MRSA and offers advantage compared to other quinolones which generally lack MRSA coverage.
• In the diabetic foot ulcer sub-group, clinical cure at TOC was high with Levonadifloxacin and also all the bacteraemic subjects in the Levonadifloxacin arm exhibited favourable clinical responses as well as microbiological success.
• The favourable efficacy of Levonadifloxacin in subjects with blood stream infection is ascribed to its high potency, rapid bactericidal action and high circulating concentrations in blood.
• Thus, Levonadifloxacin showed evidence of favourable clinical and microbiological efficacy in subjects with concurrent bacteraemia, as well as in subjects with diabetes, including diabetic foot infections caused by Gram-positive pathogens including MRSA.
• Levonadifloxacin and linezolid were both well tolerated. There were no SAEs or deaths related to study drug, and the majority of AEs were mild in severity.
• The gastrointestinal tolerability was remarkable with oral Alalevonadifloxacin and moreover no evidence for Clostridium difficile associated diarrhea was noted.
• Overall, Levonadifloxacin has undergone extensive clinical development with over 900 patients being exposed to Levonadifloxacin upto supra-therapeutic doses of 2.6 g daily and no safety concerns identified in any study.
• The results of the present study demonstrate that IV and oral Levonadifloxacin therapy are safe and well tolerated in the treatment of ABSSSI caused by Gram-positive pathogens including MRSA as well as non-inferior to IV and oral linezolid, respectively.
• Similar pharmacokinetic profile of IV and oral Levonadifloxacin provides an option for IV to oral switch for the treatment of subjects with ABSSSI.


Data of 1229 patients of any gender above 17 years of age who received Levonadifloxacin (oral or injectable) were included in the study.
A clinical diagnosis of bacterial infection was based on clinical and microbiology test results.
Data collected were recorded in a study-specific data capture tool from 177 participating sites which included clinical condition on admission, comorbidities, preexisting complications, and concomitant therapy.
Microbial testing data were collected where available and clinical therapy was administered at the discretion of a treating physician.
The study outcomes were assessed as clinical and microbiological success at the completion of therapy.
Clinical success was defined as resolution or improvement in signs and symptoms without the need of additional antimicrobial therapy, whereas persistence or worsening of signs/symptoms, the need for additional antimicrobial agents, occurrence of new infection, or death was considered clinical failure.
Microbiological success was defined as the absence of organisms at follow-up microbial testing in those patients where organisms were detected at baseline or a subsequent negative culture during a follow-up microbial testing.
Safety of treatment was assessed using the clinical and laboratory adverse events documentation, and investigators ranked therapy with Levonadifloxacin on a global assessment for efficacy and safety based on a 5-point Likert scale of excellent, very good, good, satisfactory, and poor.

Patient characteristics and pretreatment data
Of the 1229 patients, 881 (71.7%) were males and 347 (28.2%) were females and gender was not specified for one patient. The mean age was 58.51 years (ranging between 17 and 89 years).
Table 1 represents the demography, duration, and various indications for the use of Levonadifloxacin therapy in patients.
Most of the patients (n = 875, 71.2%) received IV Levonadifloxacin, whereas 303 (24.7%) patients received oral therapy and 51 (4.1%) received IV therapy followed by switchover to oral Levonadifloxacin.
Levonadifloxacin was started empirically either as monotherapy or in combination with other antimicrobial agents.
1046 (85.1%) patients were hospitalized, whereas 183 (14.9%) patients were treated on an outpatient basis, and the most common comorbid conditions were diabetes (18.3%) and hypertension (11.2%).
Other comorbidities were malignancy (2.2%), renal disorders (2.8%), ischemic heart disease (2.3%), respiratory disorders (1.6%), thyroid disorders (2.6%), and hepatic disorders (0.9%).
Lower respiratory tract infections (27.5%) and ABSSSI (18.5%) were the most common indications for the use of Levonadifloxacin.
Preexisting complications were reported in 553 (49.0%) patients, with renal impairment and septic shock being the most common complication (17.5% each), and systemic inflammatory response syndrome in 12.5% of patients.
Other preexisting complications included multi-organ failure (9.9%), hepatic impairment (5.5%), and thrombocytopenia (5.5%).
Culture report was positive in 71.8% of patients, with only 18.2% of cultures reported negative for bacterial growth.
Gram-positive infections (54.3%) were more common than Gram-negative (26.1%) and mixed (12.7%) infections
Clinical and microbiological outcome
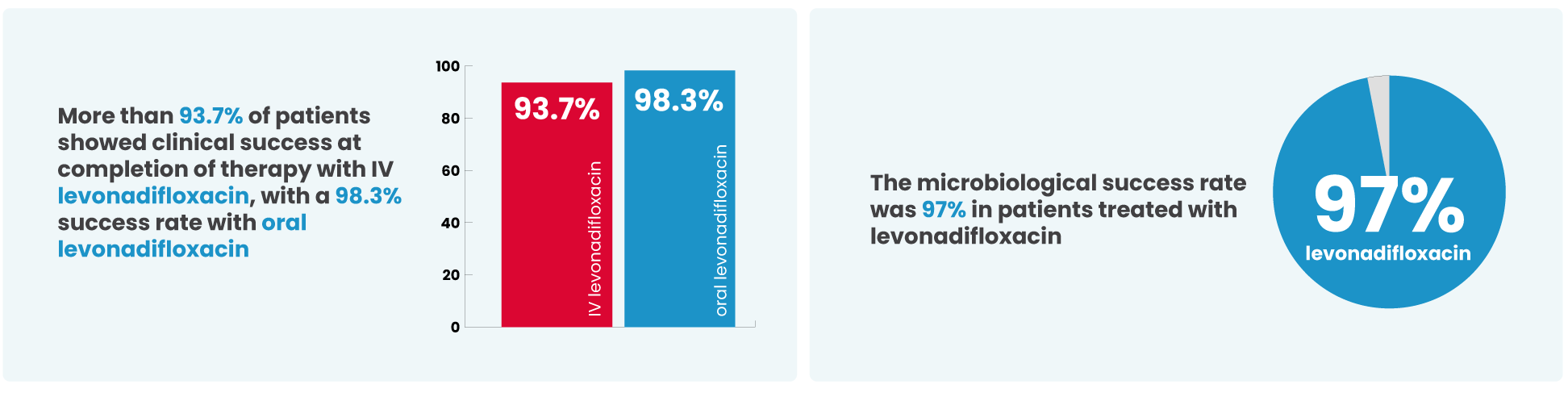
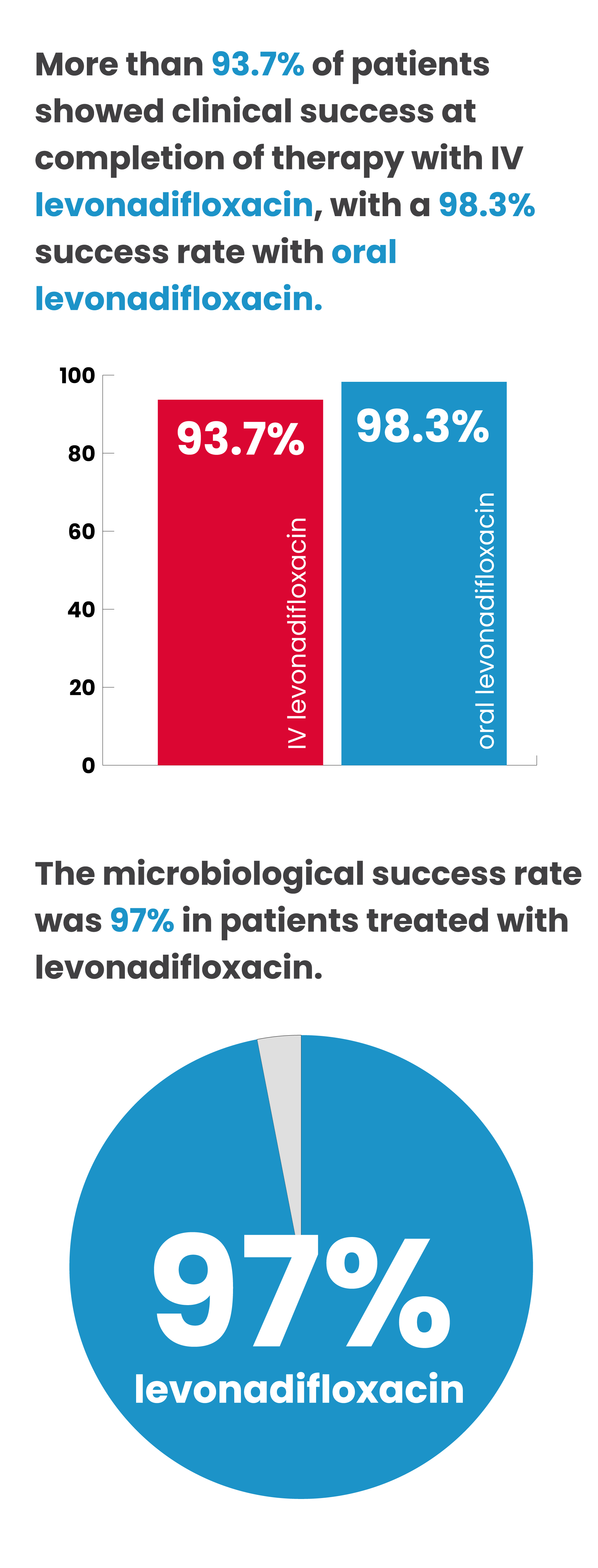
For varied indications and types of infections, the clinical success rates were from 89.3% to 100.0%.
It is noteworthy that the clinical success rates for MRSA and methicillin-susceptible S. aureus (MSSA) were 96.1% and 100.0%, respectively.
The mean time to clinical improvement was 3.89 days, 4.03 days, and 3.86 days with IV therapy, oral therapy, and IV followed by oral therapy, respectively.
The median time to improvement was 4 days (range: 1–15 days) of Levonadifloxacin therapy.
Global assessments
Overall, investigators rated the global efficacy as “good to excellent” in 96.3% of patients, and “satisfactory” in 3.7% of patients.
For global safety, investigators rated the safety as “good to excellent” in 97.3% of patients and “satisfactory” in 2.3% of patients.
Safety
There were only 11 adverse events reported in 9 patients, 2 on IV therapy and 7 on oral therapy.
The events reported were constipation (n = 2), diarrhea (n = 2), hyperglycemia (n = 1), nausea (n = 4), fatigue (n = 1), and vomiting (n = 1).
All events were of mild severity. There were no serious adverse events reported in patient records.
Indication and Uses
DOSAGE FORM AND STRENGTH
Intravenous Injection (Single use). Emrok is a clear, colourless to pale yellow, sterile, non-pyrogenic, 100 ml isotonic solution intended for intravenous infusion.
THERAPEUTIC INDICATION
Emrok is indicated in adults (≥ 18 years of age) for the treatment of Acute Bacterial Skin and Skin Structure Infections (ABSSSI) including diabetic foot infections and concurrent bacteraemia caused by susceptible isolates of the following:
Gram-positive organisms
Staphylococcus aureus (methicillin-resistant, methicillin-susceptible, quinolone-resistant, quinolone-susceptible isolates), Streptococcus pyogenes, Enterococcus faecalis, Streptococcus dysgalactiae ssp. dysgalactiae, Streptococcus agalactiae. It is critical that a Gram-negative therapy is initiated if a concomitant Gram-negative infection is suspected or documented.
Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of Emrok and other antibacterial drugs, Emrok should be used only to treat infections that are proven or strongly suspected to be caused by the susceptible strains of above listed bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
POSOLOGY
For the treatment of adults (≥ 18 years of age) with ABSSSI including diabetic foot infections, and concurrent bacteraemia; the recommended dosage regimen is as follows
- Administer 800 mg every 12 hours by intravenous infusion over a period of 90 minutes for 7-14 days or,
- Following appropriate duration of intravenous therapy, based on physician discretion, switch over to oral Alalevonadifloxacin mesylate (Emrok O) 1000 mg (two tablets of 500 mg each) every 12 hours. Emrok O tablets to be swallowed sequentially with sufficient amount of water and may be taken independent of food.
Note: For improved injection site tolerability, Emrok should be administered by intravenous infusion over a period of 90 minutes. Rapid or bolus intravenous injection or infusion of less than 90 minutes must be avoided. Emrok is not intended for intra-arterial, intramuscular, intrathecal, intraperitoneal, or subcutaneous administration.
Hepatic impairment
No dosage adjustment is required in patients with hepatic impairment (See “USE IN SPECIAL POPULATIONS” for more details)
Renal impairment
Pharmacokinetic studies with Emrok in renal impaired patients have not been conducted.
Method of Administration
Emrok is supplied in a single, ready-to-use glass bottle. The Emrok bottle should be inspected visually for particulate matter and discolouration prior to administration. Levonadifloxacin may exhibit a yellow colour that can intensify over time without adversely affecting potency. Since the bottles are for single-dose only, any unused portion remaining in the bottles should be discarded.
Emrok should be administered every 12 hours over 90 minutes by direct intravenous infusion or through a Y-type intravenous infusion set, which may already be in place. Since no data is available on the compatibility of Emrok intravenous injection with other intravenous drugs, additives or other medications should not be added to Emrok intravenous solution or infused simultaneously through the same intravenous line. If a common intravenous line is being used to administer other drugs in addition to Emrok, the line should be flushed before and after each Emrok infusion with 0.9% Sodium Chloride for Injection.
Important safety Information
CONTRAINDICATIONS
- In individuals with a known hypersensitivity to Levonadifloxacin or other quinolone antibacterials, or to any of the excipients.
- In patients with a history of tendon disorders
- In children or growing adolescents (<18 years of age)
- During pregnancy and lactation
Tendinitis and Tendon Rupture
Fluoroquinolones, including Emrok, are associated with an increased risk of tendinitis and tendon rupture in all ages and can occur within hours or weeks of starting fluoroquinolone therapy, or as long as several months after completion of fluoroquinolone therapy. This risk of developing fluoroquinolone-associated tendinitis and tendon rupture is increased in patients above 60 years of age, in patients taking corticosteroid drugs, and in patients with kidney, heart, and/or lung transplant. In the Phase III clinical study, there was no occurrence of tendinitis or tendon rupture reported in the patients treated with Emrok. At the first sign of tendon pain, swelling, or inflammation, discontinue Emrok injection, avoid exercise and use of the affected area, and inform promptly to a healthcare provider. Avoid Emrok in patients who have a history of tendon disorders or have experienced tendinitis or tendon rupture.
Peripheral Neuropathy
Fluoroquinolones have been associated with an increased risk of peripheral neuropathy. Symptoms may occur soon after initiation of fluoroquinolones and may be irreversible in some patients. In the Phase III clinical study, there was no occurrence of peripheral neuropathy reported in the patients treated with Emrok. In order to minimize the development of an irreversible condition, Emrok should be discontinued immediately if the patient experiences symptoms of peripheral neuropathy including pain, burning, tingling, numbness, and/or weakness or other alterations of sensation including light touch, pain, temperature, position sense, and vibratory sensation and/or motor strength. Avoid fluoroquinolones, including Emrok in patients who have previously experienced peripheral neuropathy.
Central Nervous System Effects
Fluoroquinolones are associated with an increased risk of central nervous system (CNS) reactions, including: convulsions, increased intracranial pressure (including pseudotumor cerebri), disturbances in attention, disorientation and toxic psychosis. Fluoroquinolones may also cause CNS reactions of nervousness, agitation, insomnia, anxiety, nightmares, paranoia, dizziness, confusion, tremors, hallucinations, depression and suicidal thoughts or acts, memory impairment, serious disturbances in mental abilities called delirium.
The mental health side effects are more prominent and more consistent across the systemic fluoroquinolone drug class. These adverse reactions may occur following the first dose. In the Phase III clinical study, there was no occurrence of any of the above mentioned drug-related reactions reported in the patients treated with Emrok. If these reactions occur in patients receiving Emrok, discontinue Emrok immediately and institute appropriate measures. As with all fluoroquinolones, use Emrok when the benefits of treatment exceed the risks in patients with known or suspected CNS disorders (e.g. severe cerebral arteriosclerosis, epilepsy) or in the presence of other risk factors that may predispose to seizures or lower the seizure threshold.
Exacerbation of Myasthenia Gravis
Fluoroquinolones have neuromuscular blocking potential and may exacerbate muscle weakness in persons with myasthenia gravis. Post-marketing serious adverse reactions, including death and requirement for ventilator support, have been associated with fluoroquinolone use in persons with myasthenia gravis. In the Phase III clinical study, no patients with myasthenia gravis were enrolled. Avoid Emrok in patients with known history of myasthenia gravis.
Hypersensitivity Reactions
Serious and occasionally fatal hypersensitive (anaphylactic) reactions, some following the first dose, have been reported in patients receiving fluoroquinolone therapy. Levonadifloxacin did not induce any hypersensitive reaction in a Guinea pig maximization test. In case of serious anaphylactic reactions, institute immediate emergency treatment with epinephrine and other resuscitative measures, including oxygen, intravenous fluids, antihistamines, corticosteroids, pressor amines, and airway management, as clinically indicated. In the Phase III clinical study there was no occurrence of a hypersensitivity reaction reported in patients treated with Emrok. Emrok should be discontinued immediately at the first appearance of a skin rash or any other sign of hypersensitivity.
Photosensitivity/Phototoxicity
Quinolones have been shown to cause photosensitivity reactions to ultraviolet (UVA and UVB) and visible radiation in patients. However, preclinical studies in Swiss mice have shown that Levonadifloxacin has a lower risk to induce photosensitivity (UVA) compared to Sparfloxacin (positive control). Moderate to severe photosensitivity/phototoxicity reactions (e.g., burning, erythema, exudation, vesicles, blistering, oedema), can be associated with the use of fluoroquinolones after sun or UV light exposure. Therefore, excessive exposure to these sources of light should be avoided. Drug therapy should be discontinued if phototoxicity occurs. In the Phase III clinical study, there was no occurrence of a photosensitivity reaction reported in patients treated with Emrok.
Clostridium difficile-Associated Diarrhoea
Clostridium difficile-associated diarrhoea (CDAD) has not been reported in any of the clinical trials of Emrok. Diarrhoea, particularly if severe, persistent and/or bloody, during or after treatment with Emrok (including several weeks after treatment), may be symptomatic of CDAD. CDAD may range in severity from mild to life threatening, the most severe form of which is pseudomembranous colitis. If CDAD is suspected or confirmed, Emrok must be stopped immediately and appropriate treatment should be initiated without delay (e.g. oral metronidazole or vancomycin). Appropriate infection control measures should be undertaken to reduce the risk of transmission. Medicinal products inhibiting the peristalsis are contraindicated in this clinical situation
Dysglycaemia
Disturbances in blood glucose, including both hypoglycaemia and hyperglycaemia have been reported with fluoroquinolones, usually in diabetic patients receiving concomitant treatment with an oral hypoglycaemic agent (for example sulphonylurea) or with insulin. In diabetic patients, careful monitoring of blood glucose is recommended. Low blood sugar levels, also called hypoglycaemia, can lead to coma. In the Phase III clinical study, there was no occurrence of hypoglycaemia and hyperglycaemia reported with Emrok O and three cases of hyperglycaemia/increased blood glucose reported with Emrok were considered to be unrelated to the study medication. If a hypoglycaemic reaction occurs, Emrok should be discontinued and appropriate therapy should be initiated immediately.
Prolongation of QT interval
In a randomized, positive-and placebo-controlled, thorough QT/QTc study conducted in US, 48 healthy subjects received Emrok O supratherapeutic dose (2600 mg), oral moxifloxacin (400 mg) and placebo. Emrok O at the supratherapeutic dose (2600 mg) did not cause any clinically significant changes on the electrocardiogram including the QTc interval.
Development of Drug-Resistant Bacteria
Prescribing Emrok in the absence of a proven or strongly suspected bacterial infections or prophylactic indication is unlikely to provide benefit to the patient and could increase the risk of the development of drug resistant bacteria.