
Prescribing Information of EMROK & EMROK O
LEVONADIFLOXACIN INJECTION (I.V.) 800mg/100ml

COMPOSITION
Levonadifiloxacin Injection (I.V.) 800mg/100ml
Each 100 ml contains:
Levonadifloxacin L-Arginine Tetrahydrate equivalent to
Levonadifloxacin ........................800 mg
Excipients:
L-Arginine I.P. ..........................................q.s.
Sodium Chloride I.P. .............................q.s.
Water for Injections I.P. .......................q.s.
DOSAGE FORM AND STRENGTHIntravenous Injection (Single use).
Emrok is a clear, colourless to pale yellow, sterile, non-pyrogenic, 100 ml isotonic
solution intended for intravenous infusion.
THERAPEUTIC INDICATION
Emrok is indicated in adults (≥18 years of age) for the treatment of Acute Bacterial Skin and Skin Structure Infections (ABSSSI) including diabetic foot infections and concurrent bacteraemia caused by susceptible isolates of the following:
Gram-positive organisms:Staphylococcus aureus (methicillin-resistant, methicillin-susceptible, quinolone-resistant, quinolone-susceptible isolates), Streptococcus pyogenes, Enterococcus faecalis, Streptococcus dysgalactiae ssp. dysgalactiae, Streptococcus agalactiae
It is critical that a Gram-negative therapy is initiated if a concomitant Gram-negative infection is suspected or documented.
UsageTo reduce the development of drug-resistant bacteria and maintain the effectiveness of Emrok and other antibacterial drugs, Emrok should be used only to treat infections that are proven or strongly suspected to be caused by the susceptible strains of above listed bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
POSOLOGY AND METHOD OF ADMINISTRATION
POSOLOGY
For the treatment of adults(≥ 18 years of age) with ABSSSI including diabetic foot infections, and concurrent bacteraemia the recommended dosage regimen is as follows
- - Administer 800 mg every 12 hours by intravenous infusion over a period of 90 minutes for 7-14 days or
- - Following appropriate duration of intravenous therapy, based on physician discretion, switch over to oral Alalevonadifloxacin mesylate (Emrok O) 1000 mg (two tablets of 500 mg each) every 12 hours. Emrok O tablets to be swallowed sequentially with suficient amount of water and may be taken independent of food.
For improved injection site tolerability, Emrok should be administered by intravenous infusion over a period of 90 minutes. Rapid or bolus intravenous injection or infusion of less than 90 minutes must be avoided. Emrok is not intended for intra-arterial, intramuscular, intrathecal, intraperitoneal, or subcutaneous administration.
Hepatic impairmentNo dosage adjustment is required in patients with hepatic impairment (See “USE IN SPECIAL POPULATIONS” for more details)
Renal impairmentPharmacokinetic studies with Emrok in renal impaired patients have not been conducted.
METHOD OF ADMINISTRATIONEmrok is supplied in a single, ready-to-use glass bottle. The Emrok bottle should be inspected visually for particulate matter and discolouration prior to administration. Levonadifloxacin may exhibit a yellow colour that can intensify over time without adversely affecting potency. Since the bottles are for single-dose only, any unused portion remaining in the bottles should be discarded.
Emrok should be administered every 12 hours over 90 minutes by direct intravenous infusion or through a Y-type intravenous infusion set, which may already be in place. Since no data is available on the compatibility of Emrok intravenous injection with other intravenous drugs, additives or other medications should not be added to Emrok intravenous solution or infused simultaneously through the same intravenous line. If a common intravenous line is being used to administer other drugs in addition to Emrok, the line should be flushed before and after each Emrok infusion with 0.9% Sodium Chloride for Injection.
CONTRAINDICATIONS- In individuals with a known hypersensitivity to Levonadifloxacin or other quinolone antibacterials, or to any of the excipients.
- In patients with a history of tendon disorders
- In children or growing adolescents (<18 years of age)
- During pregnancy and lactation
Tendinitis and Tendon Rupture
Fluoroquinolones, including Emrok, are associated with an increased risk of tendinitis and tendon rupture in all ages and can occur within hours or weeks of starting fluoroquinolone therapy, or as long as several months after completion of fluoroquinolone therapy. This risk of developing fluoroquinolone-associated tendinitis and tendon rupture is increased in patients above 60 years of age, in patients taking corticosteroid drugs, and in patients with kidney, heart, and/or lung transplant. In the Phase III clinical study, there was no occurrence of tendinitis or tendon rupture reported in the patients treated with Emrok. At the first sign of tendon pain, swelling, or inflammation, discontinue Emrok injection, avoid exercise and use of the affected area, and inform promptly to a healthcare provider. Avoid Emrok in patients who have a history of tendon disorders or have experienced tendinitis or tendon rupture.
Peripheral NeuropathyFluoroquinolones have been associated with an increased risk of peripheral neuropathy. Symptoms may occur soon after initiation of fluoroquinolones and may be irreversible in some patients. In the Phase III clinical study, there was no occurrence of peripheral neuropathy reported in the patients treated with Emrok. In order to minimize the development of an irreversible condition, Emrok should be discontinued immediately if the patient experiences symptoms of peripheral neuropathy including pain, burning, tingling, numbness, and/or weakness or other alterations of sensation including light touch, pain, temperature, position sense, and vibratory sensation and/or motor strength. Avoid fluoroquinolones, including Emrok in patients who have previously experienced peripheral neuropathy.
Central Nervous System EffectsFluoroquinolones are associated with an increased risk of central nervous system (CNS) reactions, including: convulsions, increased intracranial pressure (including pseudotumor cerebri), disturbances in attention, disorientation and toxic psychosis. Fluoroquinolones may also cause CNS reactions of nervousness, agitation, insomnia, anxiety, nightmares, paranoia, dizziness, confusion, tremors, hallucinations, depression and suicidal thoughts or acts, memory impairment, serious disturbances in mental abilities called delirium. The mental health side effects are more prominent and more consistent across the systemic fluoroquinolone drug class. These adverse reactions may occur following the first dose. In the Phase III clinical study, there was no occurrence of any of the above mentioned drug-related reactions reported in the patients treated with Emrok. If these reactions occur in patients receiving Emrok, discontinue Emrok immediately and institute appropriate measures. As with all fluoroquinolones, use Emrok when the benefits of treatment exceed the risks in patients with known or suspected CNS disorders (e.g. severe cerebral arteriosclerosis, epilepsy) or in the presence of other risk factors that may predispose to seizures or lower the seizure threshold.
Exacerbation of Myasthenia GravisFluoroquinolones have neuromuscular blocking potential and may exacerbate muscle weakness in persons with myasthenia gravis. Post-marketing serious adverse reactions, including death and requirement for ventilator support, have been associated with fluoroquinolone use in persons with myasthenia gravis. In the Phase III clinical study, no patients with myasthenia gravis were enrolled. Avoid Emrok in patients with known history of myasthenia gravis
Hypersensitivity ReactionsSerious and occasionally fatal hypersensitive (anaphylactic) reactions, some following the first dose, have been reported in patients receiving fluoroquinolone therapy. Levonadifloxacin did not induce any hypersensitive reaction in a Guinea pig maximization test. In case of serious anaphylactic reactions, institute immediate emergency treatment with epinephrine and other resuscitative measures, including oxygen, intravenous fluids, antihistamines, corticosteroids, pressor amines, and airway management, as clinically indicated. In the Phase III clinical study there was no occurrence of a hypersensitivity reaction reported in patients treated with Emrok. Emrok should be discontinued immediately at the first appearance of a skin rash or any other sign of hypersensitivity.
Photosensitivity/PhototoxicityQuinolones have been shown to cause photosensitivity reactions to ultraviolet (UVA and UVB) and visible radiation in patients. However, preclinical studies in Swiss mice have shown that Levonadifloxacin has a lower risk to induce photosensitivity (UVA) compared to Sparfloxacin (positive control). Moderate to severe photosensitivity/phototoxicity reactions (e.g., burning, erythema, exudation, vesicles, blistering, oedema), can be associated with the use of fluoroquinolones after sun or UV light exposure. Therefore, excessive exposure to these sources of light should be avoided. Drug therapy should be discontinued if phototoxicity occurs. In the Phase III clinical study, there was no occurrence of a photosensitivity reaction reported in patients treated with Emrok.
Clostridium difficile-Associated DiarrhoeaClostridium difficile-associated diarrhoea (CDAD) has not been reported in any of the clinical trials of Emrok. Diarrhoea, particularly if severe, persistent and/or bloody, during or after treatment with Emrok (including several weeks after treatment), may be symptomatic of CDAD. CDAD may range in severity from mild to life threatening, the most severe form of which is pseudomembranous colitis. If CDAD is suspected or confirmed, Emrok must be stopped immediately and appropriate treatment should be initiated without delay (e.g. oral metronidazole or vancomycin). Appropriate infection control measures should be undertaken to reduce the risk of transmission. Medicinal products inhibiting the peristalsis are contraindicated in this clinical situation.
Dysglycaemia:Disturbances in blood glucose, including both hypoglycaemia and hyperglycaemia have been reported with fluoroquinolones, usually in diabetic patients receiving concomitant treatment with an oral hypoglycaemic agent (for example sulphonylurea) or with insulin. In diabetic patients, careful monitoring of blood glucose is recommended. Low blood sugar levels, also called hypoglycaemia, can lead to coma. In the Phase III clinical study, there was no occurrence of hypoglycaemia and hyperglycaemia reported with Emrok O and three cases of hyperglycaemia/increased blood glucose reported with Emrok were considered to be unrelated to the study medication. If a hypoglycaemic reaction occurs, Emrok should be discontinued and appropriate therapy should be initiated immediately.
Prolongation of QT interval:In a randomized, positive-and placebo-controlled, thorough QT/QTc study conducted in US, 48 healthy subjects received Emrok O supratherapeutic dose (2600 mg), oral moxifloxacin (400 mg) and placebo. Emrok O at the supratherapeutic dose (2600 mg) did not cause any clinically significant changes on the electrocardiogram including the QTc interval.
Development of Drug-Resistant BacteriaPrescribing Emrok in the absence of a proven or strongly suspected bacterial infections or prophylactic indication is unlikely to provide benefit to the patient and could increase the risk of the development of drug resistant bacteria.
DRUG INTERACTIONSNo clinical drug-drug interaction studies have been conducted with Emrok or Emrok O.
Chelation Agents: Antacids, Sucralfate, Metal Cations, MultivitaminsThere are no data concerning an interaction of intravenous fluoroquinolones including Emrok with oral antacids, sucralfate, multivitamins, didanosine, or metal cations. However, Emrok should not be co-administered with any solution containing multivalent cations, e.g., magnesium, through the same intravenous line.
In Vitro Drug metabolism and Transporter studies Drug Metabolizing EnzymesIn vitro studies with cytochrome P450 (CYP) isoenzymes indicate that Levonadifloxacin and its sulphate metabolite at the concentration higher than clinical Cmax does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. In human hepatocytes, Levonadifloxacin showed no potential for in vitro induction of CYP1A2, CYP2B6 and CYP3A4/5 at concentrations 8- to 10-fold higher than clinical Cmax. The CYP450 inhibition and induction studies suggest that Levonadifloxacin is unlikely to alter the pharmacokinetics of drugs metabolized by these enzymes (e.g. midazolam, cyclosporine, warfarin, theophylline). There is a minimal CYP-mediated metabolism of Levonadifloxacin and hence drugs that are inhibitors or inducers of these enzymes are unlikely to change the pharmacokinetics of Levonadifloxacin.
TransportersIn vitro hepatic and renal transporter inhibition studies suggest that Levonadifloxacin is non-inhibitor of P-gp, BCRP OAT1, OAT3, OCT2, OATP1B1 and OATP1B3 transporters. Considering the hepatic route of excretion, in vitro substrate potentials of Levonadifloxacin was assessed using the following hepatic transporters: OATP1B1, OATP1B3, P-gp and BCRP. Levonadifloxacin is found to be a non-substrate of OATP1B1 and OATP1B3, but is a substrate of P-gp and BCRP. Based on transporter studies, the clinical drug-drug interaction due to co-administration of Levonadifloxacin with P-gp, BCRP, OAT1, OAT3, OCT2, OATP1B1 and OATP1B3 substrates or OATP1B1 and OATP1B3 inhibitors is unlikely. Drug interaction studies with P-gp or BCRP transporter inhibitors have not been evaluated clinically, the extent of change in Levonadifloxacin pharmacokinetic in the presence of these transporter inhibitors is unknown.
USE IN SPECIAL POPULATIONSPregnancy
Pregnancy Category C
The safety of use of Emrok in human pregnancy has not been evaluated. Emrok should only be used in pregnancy if indicated, i.e. only if the potential benefit outweighs the potential risk to the mother and foetus. Studies in animals indicate that Levonadifloxacin has no effect on maternal toxicity, reproduction and foetal growth. Levonadioxacin was not teratogenic in rats and rabbits at intravenous dose of 500 and 360 mg/kg/day, corresponds to approximately 7 and 10-times the highest recommended hfluman dose, respectively based upon body surface area. In a rabbit teratogenicity study, late resorption and decrease gravid uterine weight were observed at the highest dose of 360 mg/kg/day.
Nursing MothersThere are no data on the use of Emrok in nursing mothers. It is unknown whether Levonadifloxacin is excreted in human milk. In animal studies, Levonadifloxacin was detected in lactating rat milk. Based on these data it can be presumed that Levonadifloxacin will be excreted in human milk. Because of the potential for serious adverse effects in nursing infants (risk of cartilage damage based on premature animal toxicity data), a decision should be made whether to temporarily discontinue nursing or to discontinue the Emrok, taking into account the benefit of breast-feeding for the child and the benefit of therapy for the mother.
Pediatric UseThe use of Emrok in patients under 18 years of age is not recommended. Safety and effectiveness in paediatric patients below the age of 18 years have not been established. Levonadifloxacin and other quinolones have been shown to cause arthropathy in immature animals of most species. In immature dogs (4 to 5 months old), 100 mg/kg/day intravenous dose of Levonadifloxacin administered for 28 days resulted in arthropathic lesions.
Geriatric patientsGeriatric patients are at increased risk of developing severe tendon disorders including tendon rupture when treated with fluoroquinolones. This risk is further increased in patients receiving concomitant corticosteroid therapy. Caution should be used when prescribing Emrok to elderly patients especially those on corticosteroids. Patients should be informed of this potential adverse reaction and advised to discontinue Emrok and contact their healthcare providers, if any symptoms of tendinitis or tendon rupture occur.
Renal ImpairmentPharmacokinetic studies with Emrok in renal impaired patients have not been conducted.
Hepatic ImpairmentThere were no statistical significant changes observed in the plasma peak concentrations (Cmax) and area under concentration-time curve (AUC0-∞) of active parent drug Levonadifioxacin or Levonadifioxacin sulphate metabolite in patients with mild or moderate hepatic impairment (Child-Pugh Class A, or B) compared to matched healthy control subjects. Hence, dosage adjustment is not required for Emrok in mild or moderate hepatic impaired patients. In severe hepatic impaired patients (Child-Pugh Class C), there was a statistical significant (p<0.05) increase in Levonadifioxacin plasma AUC0-∞ (1.7-fold increase) compared to the matched healthy control group. Since this AUC increase was less than 2-fold, dosage adjustment is not recommended for severely hepatic impaired patients. There was no statistical significant difference in plasma exposures of Levonadifloxacin sulphate in severely hepatic impaired patients compared to matched healthy control group.
EFFECT ON ABILITY TO DRIVE AND USE MACHINEAlthough no studies on the effect of Emrok Injection on the ability to drive and use of machines have been conducted, patient may avoid operating an automobile or machinery or engage in activities such as driving as Fluoroquinolones are reported to cause dizziness, headache, visual disorders that may impair the patient's ability to concentrate and react.
UNDESIRABLE EFFECTBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure in 250 patients in the Phase III study [125 patients on Emrok (intravenous Levonadifloxacin) and 125 patients on Emrok O (Alalevonadifloxacin mesylate equivalent to Levonadifloxacin, oral tablet)]. This was a randomised active-comparator study in patients with ABSSSI. Patients were enrolled with the following infections: cellulitis/erysipelas, wound infection, major cutaneous abscess and diabetic foot infection. The baseline characteristics were comparable between the treatment groups and between the intravenous (IV) and oral subgroups. All patients were of Indian origin. Overall, patients were predominantly male (75.9% and 71.1% in the IV and oral sub-group respectively). The mean age of patients in both the IV and oral treatment groups was 45 years (range 18-65 years) and the average body mass index (BMI) ranged from 18.5 to <25 kg/m2 with 8.6% and 3.6% (IV and oral subgroups) ≥ 30 kg/m2.
The most common adverse events in patients treated with Emrok belong to Gastrointestinal Disorders (6.4 %), General Disorders and Administration Site Conditions (3.2 %), Respiratory, Thoracic and Mediastinal Disorders (2.4%), Vascular Disorders (2.4%), Renal and Urinary Disorders (1.6%), Skin and Subcutaneous Tissue Disorders (1.6%), Investigations related (1.6%) and Nervous System Disorders (1.6%).
There were four patients who discontinued from the study due to adverse events (two in the Emrok and two in IV Linezolid group). Adverse events leading to discontinuation were asphyxia (0.8%), rhonchi (0.8%) and burning sensation (0.8%). All these adverse events were assessed as not related to Emrok or Linezolid. Five serious adverse events were reported in five patients, all of which were in the IV subgroup (two in the Emrok and three in the IV Linezolid group). All these serious adverse events including deaths were considered not related to the study drug. In the Emrok group, the serious adverse events were finger amputation and asphyxia. In the Linezolid group, the serious adverse events were toe amputation, cardio-respiratory arrest, and septic shock. Among them, three patients died during the study period (one in Emrok and two in IV Linezolid group). The cause of death reported for these patients were septicaemia leading to septic shock, exacerbation of undiagnosed asthma leading to aspiration asphyxia and cardio-respiratory arrest.
Table 1: Summary of Adverse Events with Incidence (%) in Emrok + Emrok O Group by Preferred Term for the Pooled Treatment Group (Safety Population)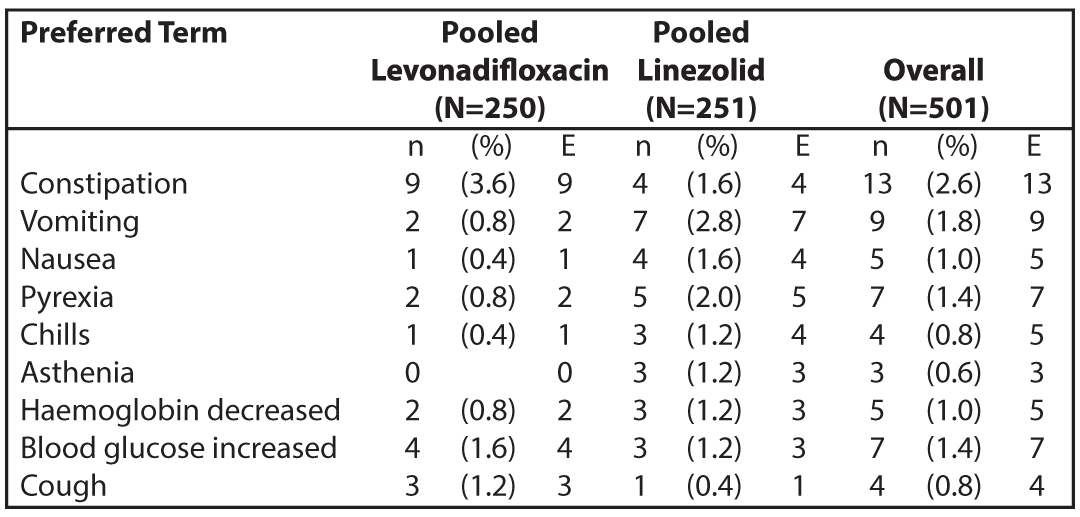
N = number of subjects at risk; n = number of subjects with TEAE; E = number of TEAEs.
OVERDOSEThe effects of an overdose of Emrok in human population are unknown. However, the highest dose of intravenous Levonadifloxacin (2.4 g/day) was found to be safe and well tolerated by healthy adult volunteers. In the event of an acute overdose, the patient should be monitored and appropriate hydration should be maintained. There is no clinical data available on the clearance of Levonadiflooxacin during haemodialysis or peritoneal dialysis.
PHARMACOLOGICAL PROPERTIESMechanism of action
Levonadifloxacin demonstrates bactericidal activity through dual inhibition of DNA gyrase and topoisomerase IV, with primary affinity towards DNA gyrase. DNA gyrase and topoisomerase IV enzymes are essential for DNA replication, transcription, repair and recombination. Owing to high affinity to DNA gyrase, Levonadifloxacin demonstrates potent cidal action even against high density Staphylococcus aureus cultures. Substitution of 4-hydroxy piperidine side chain at C-8 position of benzoquinolizine tricyclic core resulted in lower pKa (6.8), which contributes to better permeation, enhanced target affinity and a lower potential to select resistant mutants of methicillin-resistant Staphylococcus aureus (MRSA) and quinolone-resistant Staphylococcus aureus (QRSA). Levonadifloxacin due to its anionic nature demonstrates enhanced bactericidal activity against Gram-positive and Gram-negative organisms even under an acidic environment. Levonadifloxacin is not a substrate of multidrug efflux pumps, including NorA pump associated with quinolone resistance in Staphylococcus aureus.
Pharmacodynamic propertiesMechanism of resistance
Levonadifloxacin resistance can arise through mutations in defined regions of DNA gyrase or topoisomerase IV, termed the Quinolone-Resistance Determining Regions (QRDRs), or through altered effux. In vitro resistance to Levonadifloxacin develops by multiple-step mutations in the QRDRs of Gram-positive and Gram-negative bacteria. Levonadifloxacin-resistant mutants were selected in vitro at a frequency of <10-9. The mechanism of action of fluoroquinolones, including Levonadifloxacin is diflerent from that of macrolides, aminoglycosides, B-lactam, glycopeptides, tetracyclines and oxazolidinones; therefore, microorganisms resistant to these classes of drugs may still be susceptible to Levonadifloxacin. Levonadifloxacin mutant prevention concentration (MPC) for MRSA/QRSA is just 2x of minimum inhibitory concentration (MIC) demonstrating its superior resistance suppression feature.
Cross ResistanceCross resistance has been observed between Levonadifloxacin and other fluoroquinolones for Gram-negative pathogens. However, Levonadifloxacin retains potent activity against quinolone-resistant staphylococci in vitro and in in vivo animal models.
Antimicrobial ActivityLevonadifloxacin has been shown to be active against most strains of the following microorganisms, both in vitro and in clinical infections as described in the THERAPEUTIC INDICATION section.
Aerobic Gram-positive bacteria:Staphylococcus aureus (methicillin-resistant, methicillin-susceptible,
quinolone-resistant, quinolone-susceptible isolates)
Streptococcus pyogenes
Enterococcus faecalis
Streptococcus dysgalactiae ssp. dysgalactiae
Streptococcus agalactiae
Aerobic Gram-negative bacteria:Escherichia coli
Klebsiella pneumoniae
Pseudomonas aeruginosa
Acinetobacter baumannii
The following in vitro data are available but their clinical signi cance is unknown
Aerobic Gram-positive bacteriaStreptococcus pneumoniae, Streptococcus anginosus group (including S. anginosus, S. intermedius, and S. constellatus), Staphylococcus haemolyticus, Staphylococcus lugdunensis
Aerobic Gram-negative bacteria (Quinolone-susceptible strains with Levofioxacin MIC ≤2 μg/ml)Enterobacter spp., Citrobacter spp., Haemophilus influenzae, Moraxella catarrhalis, Serratia marcescens
Anaerobic Gram-positive bacteriaClostridium perfringens
Anaerobic Gram-negative bacteriaBacteroides fragilis group, Bacteroides thetaiotaomicron, Fusobacterium nucleatum, Prevotella spp., Peptostreptococcus spp.
Atypical bacteriaLegionella pneumophila, Mycoplasma pneumoniae, Mycoplasma hominis, Ureaplasma spp., Chlamydia pneumoniae
In vivo PK/PD Efficacy employing non-clinical Infection Models:Levonadifloxacin at human-simulated exposures has shown potent lung eradication effects in neutropenic murine lung infection model infected with nine Staphylococcus aureus including six MRSA (of which five were quinolone-resistant Staphylococcus aureus). Similarly, lung eradication effects of Levonadifloxacin at clinically relevant exposures in neutropenic mice have also been established against Gram-negative pathogens including E. coli, K. pneumoniae, Enterobacter, Serratia, Citrobacter and P. aeruginosa having Levonadifloxacin MIC up to 8 μg/ml. In corroboration with this, three Emrok intravenous-treated Phase III clinical trial patients had Aztreonam-resistant and Emrok-susceptible P. aeruginosa (n=2) and A. baumannii (n=1) at the baseline and were clinical responders at EOT and TOC visits.
Susceptibility Test MethodsWhen available, the clinical microbiology laboratory should provide in vitro susceptibility test results for antimicrobial drugs used in local hospitals and practice areas. Reporting these data should aid in the selection of an appropriate antibacterial drug for treatment.
Dilution TechniquesQuantitative methods are used to determine MICs. These MICs provide estimates of the susceptibility of bacteria to antimicrobial agents. The MICs should be determined using a standardized test method (broth and/or agar). The MIC values should be interpreted according to the following criteria (Table 1):
Diffusion TechniquesQuantitative methods that require measurement of zone diameters can also provide reproducible estimates of the susceptibility of bacteria to antimicrobial compounds. The zone size should be determined using a standardized test method. This procedure uses paper disks impregnated with 10 μg of Levonadifloxacin to test the susceptibility of bacteria to Levonadifloxacin. The disk diffusion breakpoints are provided in Table 2.
Table 2: Susceptibility Test Interpretive Criteria for Levonadifloxacin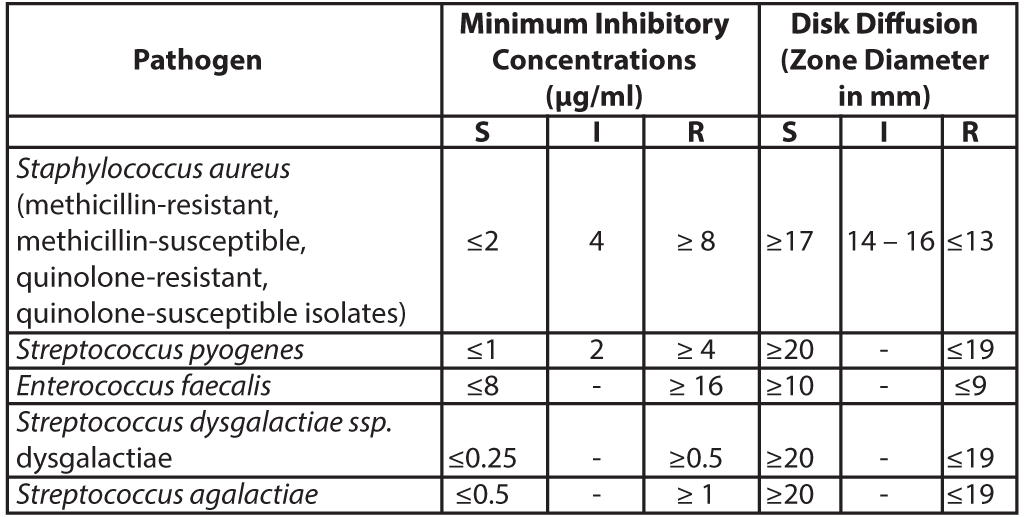
S= susceptible; I= intermediate; R= resistant
A report of Susceptible (S) indicates that the antimicrobial drug is likely to inhibit growth of the pathogen if the antimicrobial drug reaches the concentration usually achievable at the site of infection. A report of Intermediate (I) indicates that the result should be considered equivocal, and if the microorganism is not fully susceptible to alternative clinically feasible drugs, the test should be repeated. This category implies possible clinical applicability in body sites where the drug is physiologically concentrated or in situations where a high dosage of the drug can be used. This category also provides a buffer zone that prevents small uncontrolled technical factors from causing major discrepancies in interpretation. A report of Resistant (R) indicates that the antimicrobial drug is not likely to inhibit growth of the pathogen if the antimicrobial drug reaches the concentration usually achievable at the infection site; other therapy should be selected.
Quality ControlStandardized susceptibility test procedures require the use of laboratory control microorganisms (Quality control strains) to monitor and ensure the accuracy and precision of supplies and reagents used in the assay, and the techniques of the individuals performing the test. Standard Levonadifloxacin powder should provide the MIC values noted in Table 3. For the diffusion technique using the 10 μg Levonadifloxacin disk, the criteria in Table 3 should be achieved.
Table 3: Acceptable Quality Control Ranges for Levonadifloxacin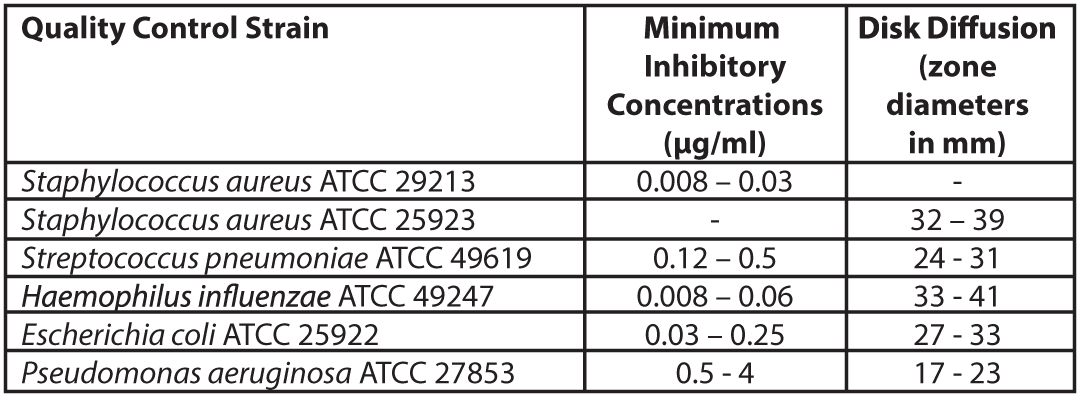 ATCC=American Type Culture Collection
Pharmacokinetic properties
ATCC=American Type Culture Collection
Pharmacokinetic properties
The mean serum pharmacokinetic parameters of Levonadifloxacin after single and multiple (every 12 hours) 800 mg intravenous infusion of Emrok in healthy male adults are summarized in the Table 4.
Table 4: Mean (standard deviation) pharmacokinetic parameters of Levonadifloxacin in healthy male adults (≥ 18 years)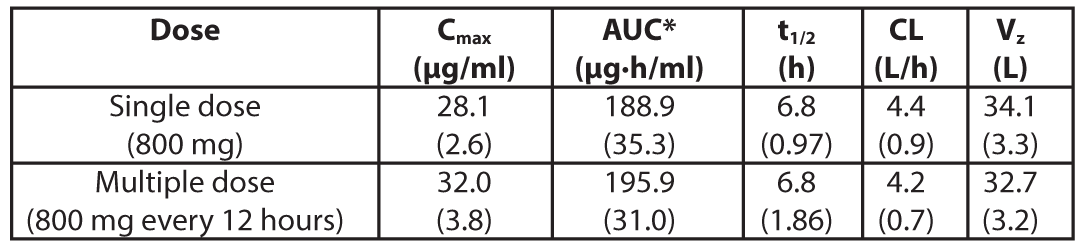
Cmax= Maximum concentration; AUC = Area under concentration-time curve; t1/2 = Elimination half-life; CL= Systemic clearance; Vz = Volume of distribution; *AUC for a single dose = AUC(0-00); for multiple dose= AUC(0-T) (AUC from time 0 to 12 hours)
Note:Oral and intravenous administration of Emrok O (Alalevonadifloxacin mesylate equivalent to Levonadifloxacin, oral tablet) and Emrok, respectively exhibited a similar pharmacokinetic profile (Cmax and AUC). This supports the option for intravenous to oral switch for the treatment of patients.
Distribution
The mean steady state volume of distribution of Levonadifloxacin is 32.7 L which approximates total body water. The serum protein binding of Levonadifloxacin is in the range of 70-90% and is independent of its concentrations in serum. Following oral administration of Emrok O (Levonadifloxacin tablets, 500 mg x 2) to healthy volunteers, the day 5 Levonadifloxacin AUC0-∞(0-12h) in lung epithelial lining fluid (ELF) (172.6 µg·h/ml) was 1.15-fold of plasma AUC(0-12h). The maximum Levonadifloxacin concentration in ELF was 26 µg/ml (Cmax)and its concentration at 12 hours was 4.28 µg/ml (Clast), resulting in 24 hours lung ELF AUC of 345.2 µg·h/ml. Intracellular concentration of Levonadifloxacin in alveolar macrophage exceeded the MIC90 values of Staphylococcus aureus and atypical respiratory pathogens. Such high exposures of Levonadifloxacin in lung would have significant therapeutic benefit for the treatment of respiratory infections.
ExcretionFollowing multiple intravenous dose administrations, the mean elimination half-life of Levonadifloxacin is approximately 6.8 hours and mean serum clearance (CL) is 4.2 L/h. The steady state is achieved on day 2. There is no evidence of accumulation of Levonadifloxacin after multiple dose administrations for a period of 5 days. After intravenous administration, approximately 16.2% of Levonadifloxacin dose is excreted as unchanged Levonadifloxacin (2.3% in urine and 13.9% in faeces) and 72.0% of Levonadifloxacin dose is excreted as Levonadifloxacin sulphate metabolite (50.3% in urine and 21.6% in faeces). A total of 88.2% of the intravenous dose is recovered as unchanged Levonadifloxacin and Levonadifloxacin sulphate metabolite in urine and faeces. Levonadifloxacin sulphate is devoid of antibacterial activity.
MetabolismLevonadifloxacin sulphate is the predominant circulating metabolite accounting for nearly half of the serum exposure (AUC) of Levonadifloxacin. Approximately 72% of intravenous Levonadifloxacin is excreted as Levonadifloxacin sulphate metabolite (approximately 50.3% in urine and 21.6% in faeces). Other Levonadifloxacin metabolites detected in trace amounts in urine are two glucuronide conjugated metabolites along with three oxidative metabolites. Trace amount of a glucuronide metabolite is also detected in faeces. The drug disposition profile of Levonadifloxacin indicates very little involvement of cytochrome P450 system in the metabolism of Levonadifloxacin.
MetabolismLevonadifloxacin sulphate is the predominant circulating metabolite accounting for nearly half of the serum exposure (AUC) of Levonadifloxacin. Approximately 72% of intravenous Levonadifloxacin is excreted as Levonadifloxacin sulphate metabolite (approximately 50.3% in urine and 21.6% in faeces). Other Levonadifloxacin metabolites detected in trace amounts in urine are two glucuronide conjugated metabolites along with three oxidative metabolites. Trace amount of a glucuronide metabolite is also detected in faeces. The drug disposition profile of Levonadifloxacin indicates very little involvement of cytochrome P450 system in the metabolism of Levonadifloxacin.
NONCLINICAL PROPERTIESANIMAL TOXICOLOGY OR PHARMACOLOGY
Levonadifloxacin exhibits a low potential for acute toxicity. Mice, rats and dogs exhibited the following clinical signs after receiving a single high dose of Levonadifloxacin: ataxia, ptosis, decreased locomotor activity, dyspnoea, prostration, tremors, and convulsions. Doses above 400 mg/kg Intravenous produced significant mortality in rodents.
Carcinogenesis, Mutagenesis, Impairment of fertilityCarcinogenesis
Long-term studies in animals to determine the carcinogenic potential of Levonadifloxacin have not been performed considering the relatively shorter duration of Emrok therapy in patients.
MutagenesisLevonadifloxacin was not mutagenic in a bacterial reverse mutation (Ames) assay, and was not clastogenic in a mouse bone marrow micronucleus test up to 475 mg/kg/day dose. In an in vitro clastogenicity assay using isolated human lymphocytes, Levonadifloxacin was negative. In a chromosomal aberration study in mice, at 475 mg/kg/day, no mutagenic effect was observed.
Impairment of fertilityLevonadifloxacin did not affect the fertility of male or female rats up to the highest intravenous dose tested (240 mg/kg/day) corresponding to approximately 3 times the recommended maximum human dose based on body surface area. Female rats were dosed 2 weeks prior to mating and through cohabitation, gestation and lactation and male rats were treated for 28 days prior to mating and 14 days during cohabitation. The serum AUC of Levonadifloxacin in male and female (non-pregnant and pregnant) rats at 240 mg/kg/day was 297 µg·h/ml
Other Non-Clinical Toxicology and Pharmacology studiesIn monkeys following a 90-minute intravenous infusion of Levonadifloxacin at a dose of 100 mg/kg (Cmax was 4.7-fold of clinical Cmax), there was no effect on the various cardiovascular parameters like systolic and diastolic blood pressure, heart rate and cardiac conduction time (PQ, QRS, QTc interval duration). In vitro studies reveal that Levonadifloxacin does not have the potential to inhibit human ether-a-go-go-related gene (hERG) potassium channels at concentrations 27-fold higher than free clinical Cmax. No seizures potentials were observed in mice administered Levonadifloxacin at supratherapeutic concentrations in combination with theophylline or fenbufen (non-steroidal anti-inflammatory drug), thus demonstrating no interaction of Levonadifloxacin with these agents.
In 28- and 90-day repeat dose IV toxicity studies with Levonadifloxacin in rat, no indication of toxicity was noticed in liver, kidney, haematology and clinical
Other Non-Clinical Toxicology and Pharmacology studiesIn monkeys following a 90-minute intravenous infusion of Levonadifloxacin at a dose of 100 mg/kg (Cmax was 4.7-fold of clinical Cmax), there was no effect on the various cardiovascular parameters like systolic and diastolic blood pressure, heart rate and cardiac conduction time (PQ, QRS, QTc interval duration). In vitro studies reveal that Levonadifloxacin does not have the potential to inhibit human ether-a-go-go-related gene (hERG) potassium channels at concentrations 27-fold higher than free clinical Cmax. No seizures potentials were observed in mice administered Levonadifloxacin at supratherapeutic concentrations in combination with theophylline or fenbufen (non-steroidal anti-inflammatory drug), thus demonstrating no interaction of Levonadifloxacin with these agents.
In 28- and 90-day repeat dose IV toxicity studies with Levonadifloxacin in rat, no indication of toxicity was noticed in liver, kidney, haematology and clinical
chemistry up to 245 and 670 mg/kg/day dose (5-10x therapeutic dose), respectively. In Intravenous 28-day dog and 90-day monkey long term studies, no evidence of any morbid or target organ toxicity was noticed at doses as high as 80 and 225 mg/kg/day, respectively (approximately 5x therapeutic dose). Dose dependent emesis was noticed in dogs which were ascribed to bolus administration of Levonadifloxacin.
CLINICAL STUDIESPhase III study in Acute Bacterial Skin and Skin Structure Infections (ABSSSI):
A total of 501 adults with clinically documented ABSSSI were enrolled in a randomised, multi-centre, open-label, non-inferiority trial comparing Emrok (800 mg) with IV Linezolid (600 mg) every 12 hours (IV subgroup) and Emrok O (1000 mg) with oral Linezolid (600 mg) every 12 hours (oral subgroup). Aztreonam 1 g every 12 hours was given to all patients for at least 3 days to cover potential Gram-negative pathogens. Treatment duration was 7 to 14 days. For patients enrolled into the IV subgroup, a switch to the respective oral therapy was allowed after at least 2 days of IV therapy. The Modified Intent-to-Treat (mITT) population included all patients who received any amount of study drug according to their randomized treatment group and had at least one post-baseline efficacy measurement.
To evaluate the treatment effect of Emrok compared with IV Linezolid, a non-inferiority analysis was conducted in 250 patients with ABSSSI (including deep/extensive cellulitis, deep abscess, or wound infection). This analysis evaluated the overall clinical cure rates at the Test-of-cure (TOC) Visit. The clinical cure rates for Emrok were numerically higher compared to Linezolid at the TOC Visit (91.0% vs. 87.8%; treatment difference: 3.2% [95%CI, -4.5 to 10.9]). The primary objective of the study was met and Emrok was non-inferior to IV Linezolid.
Table 5: Analysis of Proportion of Patients whose Overall Clinical Response Outcome was Cure at the Test of Cure (TOC) Visit (mITT Population)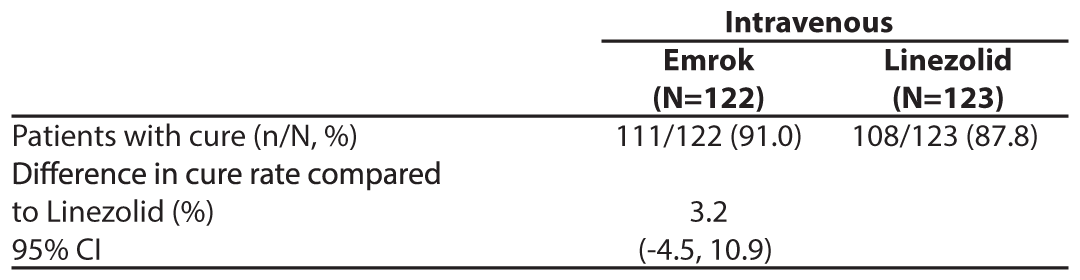
Aztreonam 1 g every 12 hours was given to all patients for at least 3 days to cover potential Gram-negative pathogens
A secondary analysis examined the proportion of patients whose clinical response outcome was responder (20% or greater reduction in lesion size compared with baseline) at Visit 3 (Day 3-4) in the mITT population (Table 6). The Responder rate was numerically higher in the Emrok group compared with the Linezolid group.
Table 6: Analysis of Proportion of Patients whose Clinical Response Outcome was Responder at Visit 3 (Day 3-4) (mITT Population)
The per-pathogen clinical cure rate was also analysed at the TOC Visit. Approximately 63% of patients had a monomicrobial infection, of which the predominant pathogen was Staphylococcus aureus with approximately 30% of patients having MRSA). In the microbiological-ITT (Micro-ITT) population, the clinical cure rate of infections due to MRSA was numerically higher for Emrok compared to Linezolid.
Table 7: Per-Pathogen Proportion of Patients with Infections due to MRSA and methicillin-susceptible Staphylococcus aureus (MSSA) with Clinical Cure at TOC Visit (Micro-ITT Population)*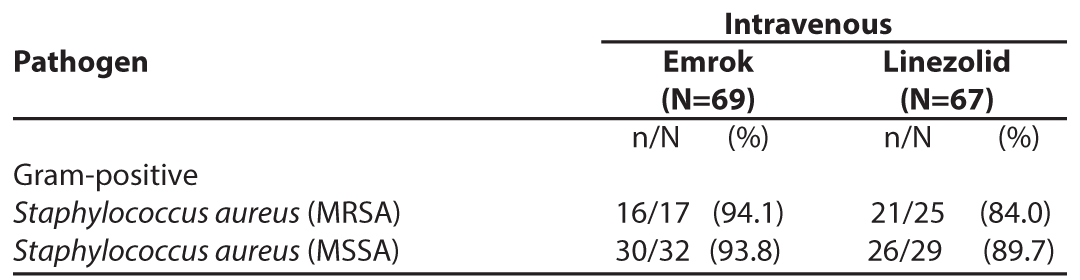
* Micro-ITT population - all patients randomized to treatment that have at least one baseline Gram-positive bacterial pathogen.
In patients with a Diabetic Foot Ulcer, the clinical cure at TOC for Emrok was numerically higher than IV Linezolid.
Table 8: Overall Clinical Response at the Test of Cure (TOC) Visit by Diabetes Status (mITT Population)
Four Emrok IV-treated subjects and two Linezolid -treated subjects (one each in IV and oral arm) had positive baseline blood cultures, indicating ABSSSI with concomitant Gram-positive bacteraemia. The four Emrok-treated subjects diagnosed with concomitant Gram-positive bacteraemia (two MRSA, one MSSA, and one S. agalactiae) at baseline were clinical responders and microbiological successes (microbiological eradication and/or presumed eradication) at the TOC Visit. The IV Linezolid-treated subject with concomitant MRSA bacteraemia at baseline, was a clinical failure despite eradication of MRSA from the blood while the oral Linezolid-treated subject was clinical responder and microbiological success.
As Emrok O has same active moiety as Emrok, results seen with Emrok are also applicable to Emrok O.
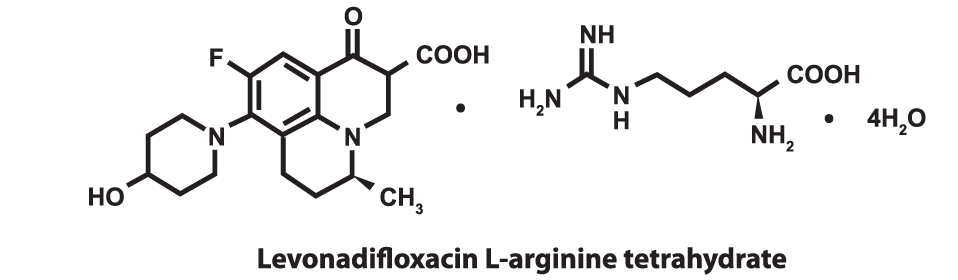
DESCRIPTIONEmrok intravenous injection contains L-arginine salt of Levonadifloxacin, a novel broad spectrum synthetic benzoquinolizine fluoroquinolone antibacterial agent. Emrok injection is for intravenous administration as a 90 minute infusion. The chemical name of Levonadifloxacin L-arginine salt is S-(-)-9-fluoro-6,7-dihy- dro-8-(4-hydroxypiperidin-1-yl)-5-methyl-1-oxo- 1H,5H-benzo[i,j] quinolizine-2-carboxylic acid L-arginine salt tetrahydrate. The molecular weight of Levonadifloxacin arginine tetrahydrate salt is 606.6g/mol and the molecular weight of Levonadifloxacin free acid is 360.4g/mol. The empirical molecular formula for Levonadifloxacin L-arginine salt is C25H 35FN6O6.4H2O and the structural formula is:
 PHARMACEUTICAL PARTICULARS
PHARMACEUTICAL PARTICULARSINCOMPATIBILITIES
Do not use any other solution other than those mentioned in ‘Method of administration’.
SHELF LIFEExpiry date is indicated on the packaging.
PACKAGING INFORMATION100 ml clear glass infusion bottle stoppered with rubber stopper and sealed with flip-off seal.
STORAGE AND HANDLING INSTRUCTIONSSPECIAL PRECAUTION FOR STORAGE
Store below 30°C (86°F) temperature and protect from light. Retain in carton until time of use. Levonadifloxacin may exhibit yellow colour that can intensify overtime without adversely affecting potency.
SPECIAL PRECAUTION FOR DISPOSAL AND HANDLINGDiscard unused portion. Disposal to be done as per local disposal guidelines.
PATIENT COUNSELING INFORMATIONInform patients that antibacterial drugs including Emrok Injection should only be used to treat bacterial infections and not viral infections (for example, the common cold). When Emrok is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance to Emrok or other antibacterial drugs in the future.
Patients should be advised that
- - They should inform the physician if they have a history of muscular disorder such as myasthenia gravis
- - They should inform the physician if they experience pain, swelling, or inflammation of a tendon, or weakness or inability to use one of their joints
- - They should inform the physician if they experience pain, burning, tingling, numbness and/or weakness of feet or hands.
- - They should inform the physician if they experience convulsions, dizziness, lightheadedness, persistent headache with or without blurred vision occurs.
- - They should inform the physician if they experience any symptoms of muscle weakness, including respiratory difficulties.
- - They should inform the physician if they experience hypersensitivity reactions, even following a single dose, and to discontinue Emrok at the first sign of a skin rash, hives or other skin reactions, a rapid heartbeat, difficulty in swallowing or breathing, any swelling suggesting angioedema (for example, swelling of the lips, tongue, face, tightness of the throat, hoarseness), or other symptoms of an allergic reaction.
- - Diarrhea is a common problem associated with antibiotic use that generally ends when the antibiotic is discontinued. Sometimes after starting treatment with antibiotics, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibiotic. If this occurs, instruct patients to contact their physician as soon as possible.
- - Inform patients particularly those with diabetes taking an oral hypoglycaemic medicine or insulin to stop the EMROK treatment, if symptoms of hypoglycaemia such as confusion, dizziness, headache, feeling shaky, sweating, pounding heart or trembling are observed and immediately contact their healthcare provider.
For reporting Adverse event / Product related issues, kindly send an email to PVSafety@wockhardt.com or contact 022-26596776.
PATENTEDDETAILS OF MANUFACTURER
MANUFACTURED IN INDIA BY :
WOCKHARDT LIMITED
E-1/1, WIDL, SEZ
Shendra MIDC Five Star Industrial Area
Shendra, Aurangabad - 431154 Maharashtra,
India.
DCGI permission no. MF-ND-150/2019 dated 30 December 2019.
DATE OF REVISION OF THE TEXT Nov 2022LEVONADIFLOXACIN TABLETS 500mg

COMPOSITION
Levonadifloxacin Tablets 500mg
Each Film Coated Tablet contains:
Alalevonadifloxacin Mesylate
equivalent to Levonadifloxacin ............500 mg
Excipients: Croscarmellose sodium I.P., hypromellose I.P., iron oxide yellow,
microcrystalline cellulose I.P., polyethylene glycol I.P., povidone I.P.,
sodium stearyl fumarate, talc I.P. and titanium dioxide I.P.
DOSAGE FORM AND STRENGTHYellow oval shaped, biconvex film coated tablet (500 mg) for oral use.
CLINICAL PARTICULARSTHERAPEUTIC INDICATION
Emrok O is indicated in adults (≥ 18 years of age) for the treatment of Acute Bacterial Skin and Skin Structure Infections (ABSSSI) including diabetic foot infections and concurrent bacteraemia caused by susceptible isolates of the following:
Gram-positive organisms:Staphylococcus aureus (methicillin-resistant, methicillin-susceptible, quinolone-resistant, quinolone-susceptible isolates), Streptococcus pyogenes, Enterococcus faecalis, Streptococcus dysgalactiae ssp. dysgalactiae, Streptococcus agalactiae
It is critical that a Gram-negative therapy is initiated if a concomitant Gram-negative infection is suspected or documented.
UsageTo reduce the development of drug-resistant bacteria and maintain the effectiveness of Emrok O and other antibacterial drugs, Emrok O should be used only to treat infections that are proven or strongly suspected to be caused by the susceptible strains of above listed bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
POSOLOGY AND METHOD OF ADMINISTRATIONPOSOLOGY
For the treatment of adults (≥ 18 years of age) with ABSSSI including diabetic foot infections and concurrent bacteraemia, the recommended dosage regimen is as follows
- - Administer 1000 mg (i.e. two 500 mg Emrok O tablets) every 12 hours by oral route for duration of 7-14 days.
- - Each 1000 mg dose comprises of two tablets of 500 mg, Emrok O tablets should be swallowed sequentially with sufficient amount of water. The tablets may be taken with or without food as food does not alter oral bioavailability.
No dosage adjustment is required in patients with hepatic impairment. (See “USE IN SPECIAL POPULATIONS” for more details)
Renal impairmentPharmacokinetic studies with Emrok O in renal impaired patients have not been conducted.
METHOD OF ADMINISTRATIONEmrok O tablets should be inspected visually for any discolouration prior to use. Emrok O tablets should be swallowed without crushing and with sufficient amount of water
CONTRAINDICATIONS- In individuals with a known hypersensitivity to Levonadifloxacin or other quinolone antibacterial, or to any of the excipients.
- In patients with a history of tendon disorders
- In children or growing adolescents (<18 years of age)
- During pregnancy and lactation
Tendinitis and Tendon Rupture
Fluoroquinolones including Emrok O are associated with an increased risk of tendinitis and tendon rupture in all ages and can occur, within hours or weeks of starting fluoroquinolone therapy, or as long as several months after completion of fluoroquinolone therapy. This risk of developing fluoroquinolone-associated tendinitis and tendon rupture is increased in patients above 60 years of age, in patients taking corticosteroid drugs, and in patients with kidney, heart, and/or lung transplant. In the Phase III clinical study, there was no occurrence of tendinitis or tendon rupture reported in the patients treated with Emrok O. At the first sign of tendon pain, swelling, or inflammation, discontinue Emrok O, avoid exercise and use of the affected area, and inform promptly to a healthcare provider. Avoid Emrok O in patients who have a history of tendon disorders or have experienced tendinitis or tendon rupture.
Peripheral NeuropathyFluoroquinolones have been associated with an increased risk of peripheral neuropathy. Symptoms may occur soon after initiation of fluoroquinolones and may be irreversible in some patients. In the Phase III clinical study, there was no occurrence of peripheral neuropathy reported in the patients treated with Emrok. In order to minimize the development of an irreversible condition, Emrok should be discontinued immediately if the patient experiences symptoms of peripheral neuropathy including pain, burning, tingling, numbness, and/or weakness or other alterations of sensation including light touch, pain, temperature, position sense, and vibratory sensation and/or motor strength. Avoid fluoroquinolones,, including Emrok in patients who have previously experienced peripheral neuropathy.
Central Nervous System EffectsFluoroquinolones are associated with an increased risk of central nervous system (CNS) reactions, including: convulsions, increased intracranial pressure (including pseudotumor cerebri), disturbances in attention, disorientation and toxic psychosis. Fluoroquinolones may also cause CNS reactions of nervousness, agitation, insomnia, anxiety, nightmares, paranoia, dizziness, confusion, tremors, hallucinations, depression and suicidal thoughts or acts, memory impairment, serious disturbances in mental abilities called delirium. The mental health side effects are more prominent and more consistent across the systemic fluoroquinolone drug class. These adverse reactions may occur following the first dose. In the Phase III clinical study, there was no occurrence of any of the above mentioned drug-related reactions reported in the patients treated with Emrok O. If these reactions occur in patients receiving Emrok O, discontinue Emrok O immediately and institute appropriate measures. As with all fluoroquinolones, use Emrok O when the benefits of treatment exceed the risks in patients with known or suspected CNS disorders (e.g. severe cerebral arteriosclerosis, epilepsy) or in the presence of other risk factors that may predispose to seizures or lower the seizure threshold.
Exacerbation of Myasthenia GravisFluoroquinolones have neuromuscular blocking potential and may exacerbate muscle weakness in persons with myasthenia gravis. Post-marketing serious adverse reactions, including death and requirement for ventilator support, have been associated with fluoroquinolone use in persons with myasthenia gravis. In the Phase III clinical study, no patients with myasthenia gravis were enrolled. Avoid Emrok O in patients with known history of myasthenia gravis.
Hypersensitivity ReactionsSerious and occasionally fatal hypersensitive (anaphylactic) reactions, some following the first dose, have been reported in patients receiving fluoroquinolone therapy. Levonadifloxacin did not induce any hypersensitive reaction in a Guinea pig maximization test. In case of serious anaphylactic reactions, institute immediate emergency treatment with epinephrine and other resuscitative measures, including oxygen, intravenous fluids, antihistamines, corticosteroids, pressor amines, and airway management, as clinically indicated. In the Phase III clinical study there was no occurrence of a hypersensitivity reaction reported in patients treated with Emrok O. Emrok O should be discontinued immediately at the first appearance of a skin rash or any other sign of hypersensitivity.
Photosensitivity/PhototoxicityQuinolones have been shown to cause photosensitivity reactions to ultraviolet (UVA and UVB) and visible radiation in patients. However, preclinical studies in Swiss mice have shown that Levonadifloxacin has a lower risk to induce photosensitivity (UVA) compared to Sparfloxacin (positive control). Moderate to severe photosensitivity/phototoxicity reactions (e.g., burning, erythema, exudation, vesicles, blistering, oedema), can be associated with the use of fluoroquinolones after sun or UV light exposure. Therefore, excessive exposure to these sources of light should be avoided. Drug therapy should be discontinued if phototoxicity occurs. In the Phase III clinical study, there was no occurrence of a photosensitivity reaction reported in patients treated with Emrok O.
Clostridium difficile-Associated DiarrhoeaClostridium difficile-associated diarrhoea (CDAD) has not been reported in any of the clinical trials of Emrok O. Diarrhoea, particularly if severe, persistent and/or bloody, during or after treatment with Emrok O (including several weeks after treatment), may be symptomatic of CDAD. CDAD may range in severity from mild to life threatening, the most severe form of which is pseudomembranous colitis. If CDAD is suspected or confirmed, Emrok O must be stopped immediately and appropriate treatment should be initiated without delay (e.g. oral metronidazole or vancomycin). Appropriate infection control measures should be undertaken to reduce the risk of transmission. Medicinal products inhibiting the peristalsis are contraindicated in this clinical situation.
Dysglycaemia:Disturbances in blood glucose, including both hypoglycemia and hyperglycemia have been reported with fluoroquinolones, usually in diabetic patients receiving concomitant treatment with an oral hypoglycemic agent (for example sulphonylurea) or with insulin. In diabetic patients, careful monitoring of blood glucose is recommended. Low blood sugar levels, also called hypoglycaemia, can lead to coma. In the Phase III clinical study, there was no occurrence of hypoglycaemia and hyperglycaemia reported with Emrok O and three cases of hyperglycaemia/increased blood glucose reported with Emrok were considered to be unrelated to the study medication. If a hypoglycaemic reaction occurs, Emrok O should be discontinued and appropriate therapy should be initiated immediately.
Prolongation of QT interval:In a randomized, positive-and placebo-controlled, thorough QT/QTc study conducted in US, 48 healthy subjects received Emrok O supratherapeutic dose (2600 mg), oral moxifloxacin (400 mg) and placebo. Emrok O at the supratherapeutic dose (2600 mg) did not cause any clinically significant changes on the electrocardiogram including the QTc interval.
Development of Drug-Resistant BacteriaPrescribing Emrok O tablets in the absence of a proven or strongly suspected bacterial infections or prophylactic indication is unlikely to provide benefit to the patient and could increase the risk of the development of drug resistant bacteria.
DRUG INTERACTIONSNo clinical drug-drug interaction studies have been conducted with Emrok or Emrok O.
Chelation Agents: Antacids, Sucralfate, Metal Cations, Multi-vitaminsAntacids containing magnesium, aluminium as well as sucralfate, metal cations such as iron and multi-vitamins preparations with zinc or didanosine may substantially interfere with the gastrointestinal absorption of Alalevonadifloxacin mesylate, resulting in considerably lower systemic levels of Levonadifloxacin (parent drug) than desired. Concomitant use of these agents, even when dosed several hours apart, should be avoided.
In Vitro Drug metabolism and Transporter studiesDrug Metabolizing Enzymes
In vitro studies with cytochrome P450 (CYP) isoenzymes indicate that Levonadifloxacin and its sulphate metabolite at the concentration higher than clinical Cmax does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. In human hepatocytes, Levonadifloxacin showed no potential for in vitro induction of CYP1A2, CYP2B6 and CYP3A4/5 at concentrations 8 to 10-fold higher than clinical Cmax. The CYP450 inhibition and induction studies suggest that Levonadifloxacin is unlikely to alter the
pharmacokinetics of drugs metabolized by these enzymes (e.g. midazolam, cyclosporine, warfarin, theophylline). There is a minimal CYP-mediated metabolism of Levonadifloxacin and hence drugs that are inhibitors or inducers of these enzymes are unlikely to change the pharmacokinetics of Levonadifloxacin.
TransportersIn vitro hepatic and renal transporter inhibition studies suggest that Levonadifloxacin is non-inhibitor of P-gp, BCRP OAT1, OAT3, OCT2, OATP1B1 and OATP1B3 transporters. Considering the hepatic route of excretion, in vitro substrate potentials of Levonadifloxacin was assessed using the following hepatic transporters: OATP1B1, OATP1B3, P-gp and BCRP. Levonadifloxacin is found to be a non-substrate of OATP1B1 and OATP1B3 but is a substrate of P-gp and BCRP. Based on transporter studies, the clinical drug-drug interaction due to co-administration of Levonadifloxacin with P-gp, BCRP, OAT1, OAT3, OCT2, OATP1B1 and OATP1B3 substrates or OATP1B1 and OATP1B3 inhibitors is unlikely. Drug interaction studies with P-gp or BCRP transporter inhibitors have not been evaluated clinically, the extent of change in Levonadifloxacin pharmacokinetic in the presence of these transporter inhibitors is unknown.
USE IN SPECIAL POPULATIONSPregnancy
Pregnancy Category C
The safety of use of Emrok O tablets in human pregnancy has not been evaluated. Emrok O tablets should only be used in pregnancy if indicated, i.e. only if the potential benefit outweighs the potential risk to the mother and foetus.
Studies in animals indicate that Levonadifloxacin has no effect on maternal toxicity, reproduction and foetal growth. Levonadifloxacin was not teratogenic in rats and rabbits at intravenous dose of 500 and 360 mg/kg/day, corresponds to approximately 7 and 10 times the highest recommended human dose, respectively based upon body surface area. In a rabbit teratogenicity study, late resorption and decrease gravid uterine weight were observed at the highest dose of 360 mg/kg/day. In pre and post-natal oral study in rats, Alalevonadifloxacin mesylate showed no effect on maternal toxicity, reproduction and growth of F2 generation up to 1500 mg/kg.
Nursing MothersThere are no data on the use of Emrok O tablets in nursing mothers. It is unknown whether Levonadifloxacin is excreted in human milk. In animal studies, Levonadifloxacin was detected in lactating rat milk. Based on these data, it can be presumed that Levonadifloxacin will be excreted in human milk. Because of the potential for serious adverse effects in nursing infants (risk of cartilage damage based on premature animal toxicity data), a decision should be made whether to temporarily discontinue nursing, or to discontinue the Emrok O tablet, taking into account the benefit of breast-feeding for the child and the benefit of therapy for mother.
Pediatric UseThe use of Emrok O in patients under 18 years of age is not recommended. Safety and effectiveness in paediatric patients below the age of 18 years have not been established.
Levonadifloxacin and other quinolones have been shown to cause arthropathy in immature animals of most species. In immature dogs (4 to 5 months old), 100 mg/kg/day intravenous dose of Levonadifloxacin administered for 28 days resulted in arthropathic lesions.
Geriatric patientsGeriatric patients are at increased risk of developing severe tendon disorders including tendon rupture when treated with fluoroquinolones. This risk is further increased in patients receiving concomitant corticosteroid therapy. Caution should be used when prescribing Emrok O tablets to elderly patients especially those on corticosteroids. Patients should be informed of this potential adverse reaction and advised to discontinue Emrok O tablets and contact their healthcare providers, if any symptoms of tendinitis or tendon rupture occur.
Renal ImpairmentPharmacokinetic studies with Emrok O in renal impaired patients have not been conducted.
Hepatic ImpairmentThere were no statistical significant changes observed in the plasma peak concentrations (Cmax) and area under concentration-time curve (AUC0-∞) of active parent drug Levonadifloxacin or Levonadifloxacin sulphate metabolite in patients with mild or moderate hepatic impairment (Child-Pugh Class A, or B) compared to matched healthy control subjects. Hence, dosage adjustment is not required for Levonadifloxacin in mild or moderate hepatic impaired patients. In severe hepatic impaired patients (Child-Pugh Class C), there was a statistical significant (p<0.05) increase in Levonadifloxacin plasma AUC0-∞ by 1.69-fold compared to the matched healthy control group. Since this AUC increase was less than 2-fold, dosage adjustment is not recommended for severely hepatic impaired patients also. There was no statistical significant difference in plasma exposures of Levonadifloxacin sulphate in severely hepatic impaired patients compared to matched healthy control group.
EFFECT ON ABILITY TO DRIVE AND USE MACHINEAlthough no studies on the effect of Emrok O on the ability to drive and use of machines have been conducted, patient may avoid operating an automobile or machinery or engage in activities such as driving as Fluoroquinolones are reported to cause dizziness, headache, visual disorders that may impair the patient's ability to concentrate and react.
UNDESIRABLE EFFECTBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure in 250 patients in the Phase III study [125 patients on Emrok (intravenous Levonadifloxacin) and 125 patients on Emrok O (Alalevonadifloxacin mesylate equivalent to Levonadifloxacin, oral tablet)]. This was a randomised active-comparator study in patients with ABSSSI. Patients were enrolled with the following infections: cellulitis/erysipelas, wound infection, major cutaneous abscess and diabetic foot infection. The baseline characteristics were comparable between the treatment groups and between the intravenous (IV) and oral subgroups. All patients were of Indian origin. Overall patients were predominantly male (75.9% and 71.1% in the IV and oral sub-group respectively). The mean age of the patients in both IV and oral treatment groups was 45 years (range 18-65 years) and the average body mass index (BMI) ranged from 18.5 to <25 kg/m2 with 8.6% and 3.6% (IV and oral subgroups) ≥ 30 kg/m2.
The duration of therapy was similar for the Emrok O and Linezolid treatment groups, with the majority of patients receiving 5-11 days of therapy in the Emrok O sub-group. Incidences of adverse events were similar between treatment groups and between IV (20.8% versus 22.4%, for Emrok and Linezolid, respectively) and oral subgroups (16.0% versus 13.5% for Emrok O and Linezolid, respectively). The overall incidence of adverse events was 18.4% in the pooled Emrok and Emrok O treatment group (IV and oral) compared with 17.9% for the pooled Linezolid group. In the Phase III study, most adverse events were reported as mild.
The most common adverse events in patients treated with Emrok O belong to Gastrointestinal Disorders (4.0%), Investigations related (3.2%), General Disorders and Administration Site Conditions (2.4%), Renal and Urinary disorders (1.6%), Skin and Subcutaneous Tissue Disorders (1.6%), Musculoskeletal and Connective Tissue Disorders (1.6%), and Nervous System Disorders (1.6%).
There were four patients who discontinued from the study due to adverse events (two in the Emrok group and two in the IV Linezolid group). Adverse events leading to discontinuation were asphyxia (0.8%), rhonchi (0.8%) and burning sensation (0.8%). All these adverse events were assessed as not related to Emrok or Linezolid. Five serious adverse events were reported in five patients, all of which were in the IV subgroup (two in the Emrok and three in the IV Linezolid group). All these serious adverse events including deaths were considered not-related to the study drug. In the Emrok group, the serious the serious adverse events were finger amputation and asphyxia. In the Linezolid group, the serious adverse events were toe amputation, cardio-respiratory arrest, and septic shock. Among them, three patients died during the study period (one in Emrok and two in IV Linezolid group). The cause of death reported for these patients. were septicaemia leading to septic shock, exacerbation of undiagnosed asthma leading to aspiration asphyxia and cardio-respiratory arrest. In the Emrok O arm, no patient discontinued study drug due to adverse event and there were no serious adverse events reported.
Table 1: Summary of Adverse Events with Incidence (%) in Emrok + Emrok O Group by Preferred Term for the Pooled Treatment Group (Safety Population)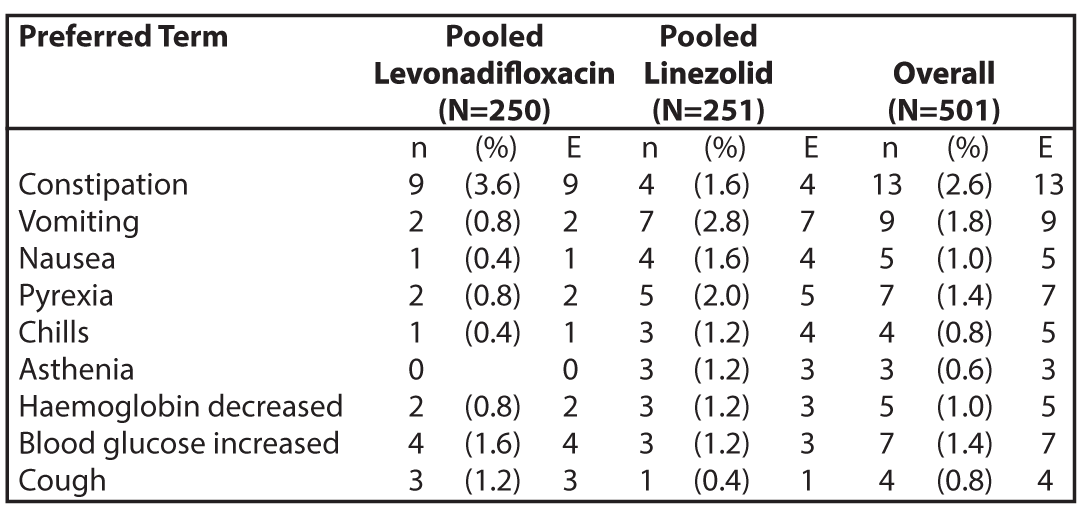
N = number of subjects at risk; n = number of subjects with TEAE; E = number of TEAEs.
OVERDOSEThe effects of an overdose of Levonadifloxacin in human population are unknown. However, the highest oral dose of Levonadifloxacin was found to be safe and well tolerated by healthy adult volunteers (2.4 g/day). In the event of an acute overdose, the stomach should be emptied. The patient should be monitored and appropriate hydration should be maintained. There is no clinical data available on the clearance of Levonadifloxacin during haemodialysis or peritoneal dialysis.
PHARMACOLOGICAL PROPERTIESMechanism of action
Levonadifloxacin demonstrates bactericidal activity through dual inhibition of DNA gyrase and topoisomerase IV, with primary affinity towards DNA gyrase. DNA gyrase and topoisomerase IV enzymes are essential for DNA replication, transcription, repair and recombination. Owing to high affinity to DNA gyrase, Levonadifloxacin demonstrates potent cidal action even against high density Staphylococcus aureus cultures. Substitution of 4-hydroxy piperidine side chain at C-8 position of benzoquinolizine tricyclic core resulted in lower pKa (6.8), which contributes to better permeation, enhanced target affinity and a lower potential to select resistant mutants of methicillin-resistant Staphylococcus aureus (MRSA) and quinolone-resistant Staphylococcus aureus (QRSA). Levonadifloxacin due to its anionic nature demonstrates enhanced bactericidal activity against Gram-positive and Gram-negative organisms even under an acidic environment. Levonadifloxacin is not a substrate of multidrug efflux pumps, including NorA pump associated with quinolone resistance in Staphylococcus aureus.
Pharmacodynamic propertiesMechanism of resistance
Levonadifloxacin resistance can arise through mutations in defined regions of DNA gyrase or topoisomerase IV, termed the Quinolone-Resistance Determining Regions (QRDRs), or through altered efflux. In vitro resistance to Levonadifloxacin develops by multiple step mutations in the QRDRs of Gram-positive and Gram-negative bacteria. Levonadifloxacin-resistant mutants were selected in vitro at a frequency of <10-9. The mechanism of action of fluoroquinolones, including Levonadifloxacin is different from that of macrolides, aminoglycosides, β-lactam, glycopeptides, tetracyclines and oxazolidinones; therefore, microorganisms resistant to these classes of drugs may still be susceptible to Levonadifloxacin and other fluoroquinolones. Levonadifloxacin mutant prevention concentration (MPC) for MRSA/QRSA is just 2x of minimum inhibitory concentration (MIC) demonstrating its superior resistance suppression feature.
Cross ResistanceCross-resistance has been observed between Levonadifloxacin and other fluoroquinolones for Gram-negative pathogens. However, Levonadifloxacin retains potent activity against quinolone-resistant staphylococci in vitro and in in vivo animal models.
Antimicrobial ActivityLevonadifloxacin has been shown to be active against most strains of the following microorganisms, both in vitro and in clinical infections as described in the THERAPEUTIC INDICATION section.
Aerobic Gram-positive bacteria:Staphylococcus aureus (methicillin-resistant, methicillin-susceptible,
quinolone-resistant, quinolone-susceptible isolates)
Streptococcus pyogenes
Enterococcus faecalis
Streptococcus dysgalactiae ssp. dysgalactiae
Streptococcus agalactiae
Aerobic Gram-negative bacteria:Escherichia coli
Klebsiella pneumoniae
Pseudomonas aeruginosa
Acinetobacter baumannii
The following in vitro data are available but their clinical significance is unknown
Aerobic Gram-positive bacteriaStreptococcus pneumoniae, Streptococcus anginosus Group (including S. anginosus, S. intermedius, and S. constellatus), Staphylococcus haemolyticus, Staphylococcus lugdunensis.
Aerobic Gram-negative bacteria (Quinolone-susceptible strains with Levofloxacin MIC ≤2 µg/mL)Enterobacter spp., Citrobacter spp., Haemophilus influenzae, Moraxella catarrhalis, Serratia marcescens.
Anaerobic Gram-positive bacteriaClostridium perfringens
Anaerobic Gram-negative bacteriaBacteroides fragilis group, Bacteroides thetaiotaomicron, Fusobacterium nucleatum, Prevotella spp., Peptostreptococcus spp.
Atypical bacteriaLegionella pneumophila, Mycoplasma pneumoniae, Mycoplasma hominis, Ureaplasma spp., Chlamydia pneumoniae
In vivo PK/PD Efficacy employing non-clinical Infection ModelsLevonadifloxacin at human-simulated exposures has shown potent lung eradication effects in neutropenic murine lung infection model infected with nine Staphylococcus aureus including six MRSA (of which 5 were quinolone-resistant Staphylococcus aureus). Similarly, lung eradication effects of Levonadifloxacin at clinically relevant exposures in neutropenic mice have also been established against Gram-negative pathogens including E. coli, K. pneumoniae, Enterobacter, Serratia, Citrobacter and P. aeruginosa having Levonadifloxacin MIC up to 8 µg/ml. In corroboration with this, two Emrok O treated patients in the Phase III clinical trial had Aztreonam-resistant and Emrok-susceptible E. coli and A. baumannii at the baseline and were clinical responders at EOT and TOC visits.
Susceptibility Test MethodsWhen available, the clinical microbiology laboratory should provide in vitro susceptibility test results for antimicrobial drugs used in local hospitals and practice areas. Reporting these data should aid in the selection of an appropriate antibacterial drug for treatment.
Dilution TechniquesQuantitative methods are used to determine MICs. These MICs provide estimates of the susceptibility of bacteria to antimicrobial agents. The MICs should be determined using a standardized test method (broth and/or agar). The MIC values should be interpreted according to the following criteria (Table 2):
Diffusion TechniquesQuantitative methods that require measurement of zone diameters can also provide reproducible estimates of the susceptibility of bacteria to antimicrobial compounds. The zone size should be determined using a standardized test method. This procedure uses paper disks impregnated with 10 µg of Levonadifloxacin to test the susceptibility of bacteria to Levonadifloxacin. The disk diffusion breakpoints are provided in Table 2.
Table 2: Susceptibility Test Interpretive Criteria for Levonadifloxacin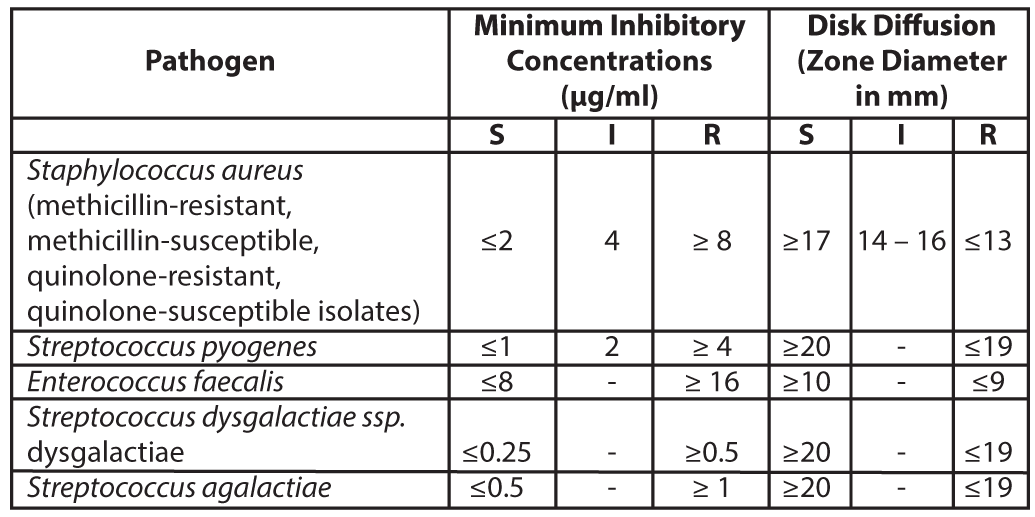
S = susceptible, I= Intermediate, R = resistant
A report of Susceptible (S) indicates that the antimicrobial drug is likely to inhibit growth of the pathogen if the antimicrobial drug reaches the concentration usually achievable at the site of infection. A report of Intermediate (I) indicates that the result should be considered equivocal, and if the microorganism is not fully susceptible to alternative clinically feasible drugs, the test should be repeated. This category implies possible clinical applicability in body sites where the drug is physiologically concentrated or in situations where a high dosage of the drug can be used. This category also provides a buffer zone that prevents small uncontrolled technical factors from causing major discrepancies in interpretation. A report of Resistant (R) indicates that the antimicrobial drug is not likely to inhibit growth of the pathogen if the antimicrobial drug reaches the concentration usually achievable at the infection site; other therapy should be selected.
Quality ControlStandardized susceptibility test procedures require the use of laboratory control microorganisms (Quality control strains) to monitor and ensure the accuracy and precision of supplies and reagents used in the assay, and the techniques of the individuals performing the test. Standard Levonadifloxacin powder should provide the MIC values noted in Table 3. For the diffusion technique using the 10 µg Levonadifloxacin disk, the criteria in Table 3 should be achieved.
Table 3: Acceptable Quality Control Ranges for Levonadifloxacin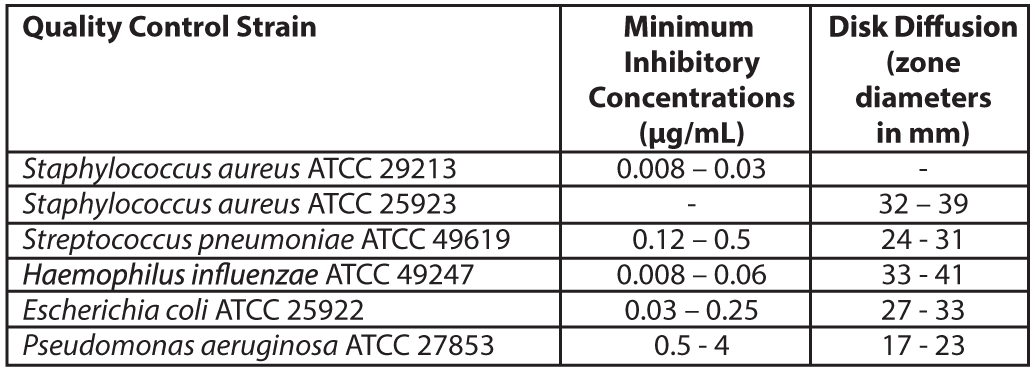 Pharmacokinetic Properties
Pharmacokinetic PropertiesThe mean serum pharmacokinetic parameters of active drug Levonadifloxacin after single and multiple (every 12 hours) 1000 mg oral dose of Emrok O tablets in healthy male adults are summarized in the Table 4.
Table 4: Mean pharmacokinetic parameters of Levonadifloxacin in healthy male adults (≥18 years)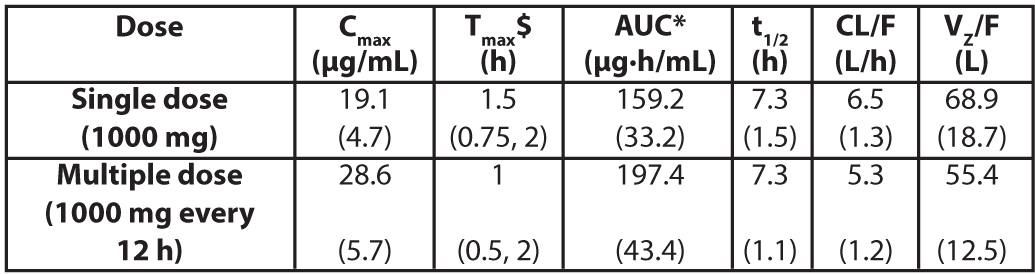
Values in parenthesis are standard deviation; $ value represented as median (range) Cmax= Maximum concentration; Tmax= Time to reach Cmax, AUC = Area under concentration-time curve; t1/2 = Elimination half-life; CL/F = Apparent clearance after oral administration; Vz/F = Apparent volume of distribution after oral administration; *AUC for single dose = AUC(0-∞); for multiple dose = AUC(0-τ) (AUC from time 0 to 12 hours)
AbsorptionFollowing oral administration, Alalevonadifloxacin mesylate undergoes esterase mediated bio-transformation to release active drug Levonadifloxacin in systemic circulation. Prodrug Alalevonadifloxacin mesylate does not appear in systemic circulation after oral administration. As esterases are widely distributed in diverse tissues such as intestinal mucosa, liver and blood, there is a rapid and complete conversion of prodrug into Levonadifloxacin. The day 5 serum concentration time profile of Levonadifloxacin after oral multiple doses of 1000 mg was almost comparable to that of Levonadifloxacin 800 mg intravenous infusion (Figure 1). The absolute oral bioavailability of Levonadifloxacin was approximately 90 % under fasting condition and the peak serum concentrations of Levonadifloxacin were usually attained at 0.5 to 2 hours after tablet administrations suggesting a rapid and almost complete oral absorption of Alalevonadifloxacin mesylate. Administration of Emrok O tablets (1000 mg) following high-calorie-high-fat diet delayed the time of Levonadifloxacin to achieve peak concentration (Tmax) by 2 hours and decreased Levonadifloxacin peak concentration (Cmax) by approximately 27% without significantly affecting the area under the plasma concentration-time curve (AUC) values. Food has little effect on the oral absorption of Alalevonadifloxacin mesylate.
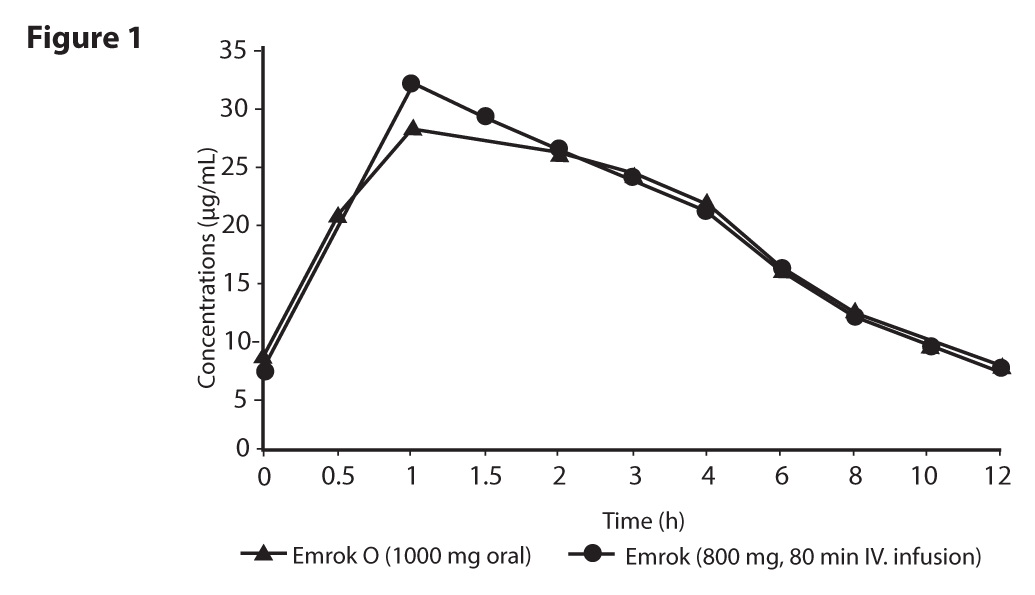 Distribution
Distribution
The mean steady state apparent volume of distribution of Levonadifloxacin after oral administration is 55.44 L which approximates total body water. The serum protein binding of Levonadifloxacin is in the range of 70-90% and is independent of its concentrations in serum. Following oral administration of Emrok O Tablet (Levonadifloxacin tablets, 500 mg × 2) to healthy volunteers, the day 5 Levonadifloxacin AUC(0-12h) in lung epithelial lining fluid (ELF) (172.6 µg·h/mL) was 1.15-fold of plasma AUC(0-12h). The maximum Levonadifloxacin concentration in ELF was 26 µg/mL (Cmax) and its concentration at 12 hours was 4.28 µg/mL (Clast), resulting in to 24 hours lung ELF AUC of 345.2 µg·h/mL. Intracellular concentration of Levonadifloxacin in alveolar macrophage exceeded the MIC90 values of Staphylococcus aureus and atypical respiratory pathogens. Such high exposures of Levonadifloxacin in lung would have significant therapeutic benefit for the treatment of respiratory infections.
MetabolismLevonadifloxacin sulphate is the predominant circulating metabolite accounting for nearly half of the serum exposure (AUC) of Levonadifloxacin. Approximately, 66.7% of oral total dose of Levonadifloxacin is excreted as Levonadifloxacin sulphate metabolite. Significant portion of Levonadifloxacin sulphate metabolite is excreted through urine (37.2% of the dose) and faeces (29.5% of dose). Other metabolites of Levonadifloxacin detected in trace amount in urine are two glucuronide conjugated metabolites along with three oxidative metabolites. Trace amount of a glucuronide metabolite is also detected in faeces. The drug disposition profile of Levonadifloxacin indicates very little involvement of cytochrome P450 system in the metabolism of Levonadifloxacin.
NONCLINICAL PROPERTIESANIMAL TOXICOLOGY OR PHARMACOLOGY
Alalevonadifloxacin mesylate exhibits a low potential for acute toxicity. Mice, rats and dogs exhibited the following clinical signs after receiving a single high dose of Alalevonadifloxacin mesylate, ptosis, decreased locomotor activity, dysponea, prostration, and convulsions. Doses above 4500 mg/kg oral in rats and above 2400 mg/kg oral in mice produced significant mortality in rodents.
Carcinogenesis, Mutagenesis, Impairment of fertilityCarcinogenesis
Long-term studies in animals to determine the carcinogenic potential of Levonadifloxacin have not been performed considering the relatively shorter duration of Emrok O therapy in patients.
MutagenesisAlalevonadifloxacin mesylate was not mutagenic in a bacterial reverse mutation (Ames) assay, and was not clastogenic in a mouse bone marrow micronucleus test up to 1500 mg/kg/day dose. In an in vitro clastogenicity assay using isolated human lymphocytes, Alalevonadifloxacin mesylate was negative. In a chromosomal aberration study in rat, at 1200 mg/kg/day no mutagenic effect was observed.
Impairment of fertilityLevonadifloxacin had no effect on fertility in male or female rats at an intravenous dose of 240 mg/kg/day, approximately 3 times the maximum recommended human intravenous dose based on body surface area. Female rats were dosed 2 weeks prior to mating and through cohabitation, gestation and lactation and male rats were treated for 28 days prior to mating and 14 days during cohabitation. The serum AUC of Levonadifloxacin in male and female (non-pregnant and pregnant) rats at 240 mg/kg/day was 297 µg·h/mL.
Other Non-Clinical Toxicology and Pharmacology studiesIn monkeys following a 90-minute intravenous infusion of Levonadifloxacin at a dose of 100 mg/kg (Cmaxwas 4.7-fold of clinical Cmax ), there was no effect on the various cardiovascular parameters like systolic and diastolic blood pressure, heart rate and cardiac conduction time (PQ, QRS, QTc interval duration). In vitro studies reveal that Levonadifloxacin does not have the potentials to inhibit human ether-a-go-go-related gene (hERG) potassium channels at concentrations 27-fold higher than free clinical Cmax . No seizures potentials were observed in mice administered Levonadifloxacin at supratherapeutic concentrations in combination with theophylline or fenbufen (non-steroidal anti-inflammatory drug), thus demonstrating no interaction of Levonadifloxacin with these agents.
The no-observed-adverse-effect level (NOAEL) for Alalevonadifloxacin mesylate in a subacute 4-week oral study in Wistar rats was found to be 1000 mg/kg/day. The corresponding NOAEL for a 70 kg human would be 11.7 g/day. The NOAEL for Alalevonadifloxacin mesylate in a subchronic 13-week oral study in Wistar rats and Cyanomolgus monkey was found to be 1400 and 336 mg/kg/day, respectively. The corresponding human dose based on the NOAEL in rat and monkey is 15 g and 7.5 g per day for 70 kg human, respectively.
CLINICAL STUDIES Phase III study in Acute Bacterial Skin and Skin Structure Infections (ABSSSI):A total of 501 adults with clinically documented ABSSSI were enrolled in a randomised, multi-centre, open-label, non-inferiority trial comparing Emrok (800 mg) with IV Linezolid (600 mg) every 12 hours (IV subgroup) and Emrok O (1000 mg) with oral Linezolid (600 mg) every 12 hours (oral subgroup). Aztreonam 1 g every 12 hours was given to all patients for at least 3 days to cover potential Gram-negative pathogens. Treatment duration was 7 to 14 days. For patients enrolled into the IV subgroup, a switch to the respective oral therapy was allowed after at least 2 days of IV therapy. The Modified Intent-to-Treat (mITT) population included all patients who received any amount of study drug according to their randomized treatment group and had at least one post-baseline efficacy measurement. To evaluate the treatment effect of Emrok O compared with oral Linezolid, a non-inferiority analysis was conducted in 251 patients with ABSSSI (including deep/extensive cellulitis, deep abscess, or a wound infection). This analysis evaluated the overall clinical cure rates at the Test-of-cure (TOC) Visit.
The clinical cure rates of Emrok O were numerically higher compared to Linezolid at TOC Visit (95.2% vs. 93.6%; treatment difference of 1.6 % [95%CI, -4.2 to 7.3]). The primary objective of the study was met and Emrok O was non-inferior to oral Linezolid.
Table 5: Analysis of Proportion of Patients whose Overall Clinical Response Outcome was Cure at the Test of Cure (TOC) Visit (mITT Population)
Aztreonam 1 g every 12 hours was given to all patients for at least 3 days to cover potential Gram-negative pathogens
A secondary analysis examined the proportion of patients whose clinical response outcome was responder (20% or greater reduction in lesion size compared with baseline) at Visit 3 (Day 3-4) in the mITT population (Table 6).The responder rate was numerically higher in the Emrok O group compared with Linezolid group.
Table 6: Analysis of Proportion of Patients whose Clinical Response Outcome was Responder at Visit 3 (Day 3-4) (mITT Population)
The per-pathogen clinical cure rate was also analysed at the TOC Visit. Approximately 63% of patients had a monomicrobial infection, of which the predominant pathogen was Staphylococcus aureus with approximately 30% of patients having MRSA). In the microbiological-ITT (Micro-ITT) population, the clinical cure rate of infections due to MRSA was numerically higher for Emrok O compared to the oral Linezolid.
Table 7: Per-Pathogen Proportion of Patients with Infection due to MRSA and methicillin-susceptible Staphylococcus aureus (MSSA) with Clinical Cure at TOC Visit (Micro-ITT Population*)
* Micro-ITT population - all patients randomized to treatment that have at least one baseline Gram-positive bacterial pathogen.
In the Diabetic Foot Ulcer sub-group, clinical cure at TOC for Emrok O and oral Linezolid was high and similar.
Table 7: Per-Pathogen Proportion of Patients with Infection due to MRSA and methicillin-susceptible Staphylococcus aureus (MSSA) with Clinical Cure at TOC Visit (Micro-ITT Population*)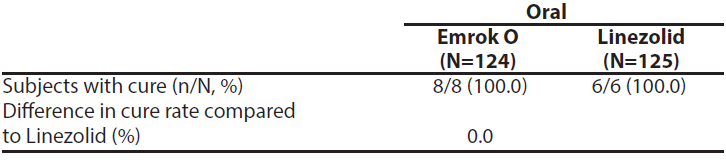
Four Emrok IV-treated subjects and two Linezolid -treated subjects (one each in IV and oral arm) had positive baseline blood cultures, indicating ABSSSI with concomitant Gram-positive bacteraemia. The four Emrok-treated subjects diagnosed with concomitant Gram-positive bacteraemia (two MRSA, one MSSA, and one S. agalactiae) at baseline were clinical responders and microbiological successes (microbiological eradication and/or presumed eradication) at the TOC Visit. The IV Linezolid-treated subject with concomitant MRSA bacteraemia at baseline, was a clinical failure despite eradication of MRSA from the blood while the oral Linezolid-treated subject was clinical responder and microbiological success.
As Emrok O has same active moiety as Emrok, results seen with Emrok are also applicable to Emrok O.
DESCRIPTIONEmrok O (Alalevonadifloxacin mesylate; prodrug of Levonadifloxacin) is a mesylate salt of L-alanine ester of the active parent drug Levonadifloxacin, a synthetic broad spectrum benzoquinolizine fluoroquinolone antibacterial agent for oral administration. Alalevonadifloxacin is rapidly and completely hydrolysed by esterase enzymes during oral absorption, and is distributed in the circulating blood as active Levonadifloxacin. The chemical name of Alalevonadifloxacin mesylate is (S)-(-)-9-Fluoro-8-(4-L-alaninyl oxypiperidin-1-yl)-5-methyl-6, 7-dihydro-1-oxo-1H,5H-benzo[i,j] quinolizine-2-carboxylic acid, methane sulfonic acid salt and its structure is shown below. The molecular formula of Alalevonadifloxacin mesylate is C22H26FN3O5•CH3SO3H and the molecular weight is 527.6 g/mol. Alalevonadifloxacin mesylate is freely soluble in water at 25°C across a pH range of 2-8 encountered in the gastrointestinal tract. The molecular weight of Levonadifloxacin free acid is 360.4 g/mol.
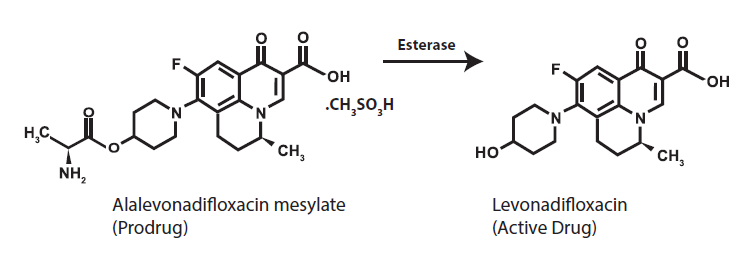 PHARMACEUTICAL PARTICULARS
PHARMACEUTICAL PARTICULARSINCOMPATIBILITIES
Not Applicable
SHELF LIFEExpiry date is indicated on the packaging.
PACKAGING INFORMATIONEmrok O Tablets: Blister pack of 4 (2x2) tablets
STORAGE AND HANDLING INSTRUCTIONSSPECIAL PRECAUTION FOR STORAGE
Store protected from moisture at a temperature not exceeding 30°C. Keep out of reach of children
PATIENT COUNSELING INFORMATIONInform patients that antibacterial drugs including Emrok O Tablets should only be used to treat bacterial infections and not viral infections (for example, the common cold). When Emrok O Tablet is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance to Emrok O or other antibacterial drugs in the future.
Patients should be advised that
- - Emrok O Tablets may be taken with or without food and without any dietary restrictions
- - Emrok O Tablets should be taken at least 2 hours before or 6 hours after antacids containing magnesium, or aluminium, with sucralfate, with metal cations such as iron, or with multivitamin preparations containing calcium, zinc or iron, or with didanosine buffered tablets for oral suspension.
- - They should inform the physician if they have a history of muscular disorder such as myasthenia gravis
- - They should inform the physician if they experience pain, swelling, or inflammation of a tendon, or weakness or inability to use one of their joints
- - They should inform the physician if they experience pain, burning, tingling, numbness and/or weakness of feet or hands.
- - They should inform the physician if they experience convulsions, dizziness, lightheadedness, persistent headache with or without blurred vision occurs.
- - They should inform the physician if they experience any symptoms of muscle weakness, including respiratory difficulties.
- - They should inform the physician if they experience hypersensitivity reactions, even following a single dose, and to discontinue Emrok O at the first sign of a skin rash, hives or other skin reactions, a rapid heartbeat, difficulty in swallowing or breathing, any swelling suggesting angioedema (for example, swelling of the lips, tongue, face, tightness of the throat, hoarseness), or other symptoms of an allergic reaction.
- - Diarrhea is a common problem associated with antibiotic use that generally ends when the antibiotic is discontinued. Sometimes after starting treatment with antibiotics, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibiotic. If this occurs, instruct patients to contact their physician as soon as possible
- - Inform patients particularly those with diabetes taking an oral hypoglycaemic medicine or insulin to stop the Emrok O treatment, if symptoms of hypoglycaemia such as confusion, dizziness, headache, feeling shaky, sweating, pounding heart or trembling are observed and immediately contact their healthcare provider.
For reporting Adverse event / Product related issues, kindly send an email to PVSafety@wockhardt.com or contact 022-26596776.
PATENTEDDETAILS OF MANUFACTURER
MANUFACTURED IN INDIA BY :
WOCKHARDT LIMITED E-1/1, WIDL, SEZ
Shendra MIDC Five Star Industrial Area
Shendra, Aurangabad - 431154
Maharashtra, India.
DETAILS OF PERMISSION
DCGI permission no. MF-ND-150/2019 dated 30 December 2019.
DATE OF REVISION OF THE TEXT
Nov 2022